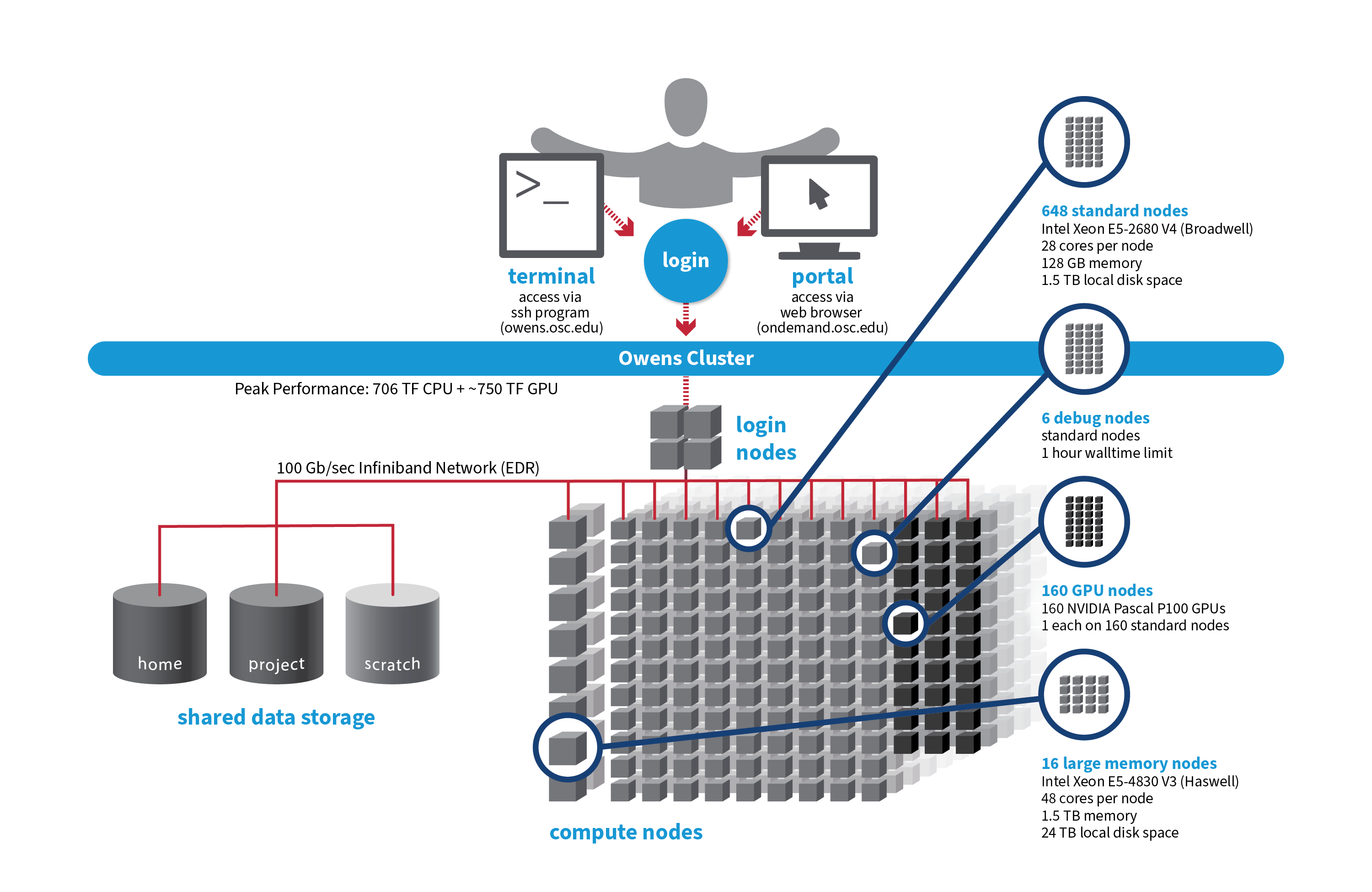
Owens
OSC's Owens cluster being installed in 2016 is a Dell-built, Intel® Xeon® processor-based supercomputer.
Hardware

Detailed system specifications:
Please see owens batch limits for details on user available memory amount.
- 824 Dell Nodes
- Dense Compute
-
648 compute nodes (Dell PowerEdge C6320 two-socket servers with Intel Xeon E5-2680 v4 (Broadwell, 14 cores, 2.40 GHz) processors, 128 GB memory)
-
-
GPU Compute
-
160 ‘GPU ready’ compute nodes -- Dell PowerEdge R730 two-socket servers with Intel Xeon E5-2680 v4 (Broadwell, 14 cores, 2.40 GHz) processors, 128 GB memory
-
NVIDIA Tesla P100 (Pascal) GPUs -- 5.3 TF peak (double precision), 16 GB memory
-
-
Analytics
-
16 huge memory nodes (Dell PowerEdge R930 four-socket server with Intel Xeon E5-4830 v3 (Haswell 12 core, 2.10 GHz) processors, 1,536 GB memory, 12 x 2 TB drives)
-
- 23,392 total cores
- 28 cores/node & 128 GB of memory/node
- Mellanox EDR (100 Gbps) Infiniband networking
- Theoretical system peak performance
- ~750 teraflops (CPU only)
- 4 login nodes:
- Intel Xeon E5-2680 (Broadwell) CPUs
- 28 cores/node and 256 GB of memory/node
- IP address: 192.148.247.[141-144]
How to Connect
-
SSH Method
To login to Owens at OSC, ssh to the following hostname:
owens.osc.edu
You can either use an ssh client application or execute ssh on the command line in a terminal window as follows:
ssh <username>@owens.osc.edu
You may see warning message including SSH key fingerprint. Verify that the fingerprint in the message matches one of the SSH key fingerprint listed here, then type yes.
From there, you are connected to Owens login node and have access to the compilers and other software development tools. You can run programs interactively or through batch requests. We use control groups on login nodes to keep the login nodes stable. Please use batch jobs for any compute-intensive or memory-intensive work. See the following sections for details.
-
OnDemand Method
You can also login to Owens at OSC with our OnDemand tool. The first step is to login to OnDemand. Then once logged in you can access Owens by clicking on "Clusters", and then selecting ">_Owens Shell Access".
Instructions on how to connect to OnDemand can be found at the OnDemand documention page.
File Systems
Owens accesses the same OSC mass storage environment as our other clusters. Therefore, users have the same home directory as on the old clusters. Full details of the storage environment are available in our storage environment guide.
Software Environment
The module system is used to manage the software environment on owens. Use module load <package> to add a software package to your environment. Use module list to see what modules are currently loaded and module avail to see the modules that are available to load. To search for modules that may not be visible due to dependencies or conflicts, use module spider . By default, you will have the batch scheduling software modules, the Intel compiler and an appropriate version of mvapich2 loaded.
You can keep up to on the software packages that have been made available on Owens by viewing the Software by System page and selecting the Owens system.
Compiling Code to Use Advanced Vector Extensions (AVX2)
The Haswell and Broadwell processors that make up Owens support the Advanced Vector Extensions (AVX2) instruction set, but you must set the correct compiler flags to take advantage of it. AVX2 has the potential to speed up your code by a factor of 4 or more, depending on the compiler and options you would otherwise use.
In our experience, the Intel and PGI compilers do a much better job than the gnu compilers at optimizing HPC code.
With the Intel compilers, use -xHost and -O2 or higher. With the gnu compilers, use -march=native and -O3 . The PGI compilers by default use the highest available instruction set, so no additional flags are necessary.
This advice assumes that you are building and running your code on Owens. The executables will not be portable. Of course, any highly optimized builds, such as those employing the options above, should be thoroughly validated for correctness.
See the Owens Programming Environment page for details.
Batch Specifics
Refer to the documentation for our batch environment to understand how to use the batch system on OSC hardware. Some specifics you will need to know to create well-formed batch scripts:
- Most compute nodes on Owens have 28 cores/processors per node. Huge-memory (analytics) nodes have 48 cores/processors per node.
- Jobs on Owens may request partial nodes.
Using OSC Resources
For more information about how to use OSC resources, please see our guide on batch processing at OSC. For specific information about modules and file storage, please see the Batch Execution Environment page.
Technical Specifications
The following are technical specifications for Owens.
- Number of Nodes
-
824 nodes
- Number of CPU Sockets
-
1,648 (2 sockets/node)
- Number of CPU Cores
-
23,392 (28 cores/node)
- Cores Per Node
-
28 cores/node (48 cores/node for Huge Mem Nodes)
- Local Disk Space Per Node
-
~1,500GB in /tmp
- Compute CPU Specifications
-
Intel Xeon E5-2680 v4 (Broadwell) for compute
- 2.4 GHz
- 14 cores per processor
- Computer Server Specifications
-
- 648 Dell PowerEdge C6320
- 160 Dell PowerEdge R730 (for accelerator nodes)
- Accelerator Specifications
-
NVIDIA P100 "Pascal" GPUs 16GB memory
- Number of Accelerator Nodes
-
160 total
- Total Memory
- ~ 127 TB
- Memory Per Node
-
128 GB (1.5 TB for Huge Mem Nodes)
- Memory Per Core
-
4.5 GB (31 GB for Huge Mem)
- Interconnect
-
Mellanox EDR Infiniband Networking (100Gbps)
- Login Specifications
-
4 Intel Xeon E5-2680 (Broadwell) CPUs
- 28 cores/node and 256GB of memory/node
- Special Nodes
-
16 Huge Memory Nodes
- Dell PowerEdge R930
- 4 Intel Xeon E5-4830 v3 (Haswell)
- 12 Cores
- 2.1 GHz
- 48 cores (12 cores/CPU)
- 1.5 TB Memory
- 12 x 2 TB Drive (20TB usable)
Environment changes in Slurm migration
As we migrate to Slurm from Torque/Moab, there will be necessary software environment changes.
Decommissioning old MVAPICH2 versions
Old MVAPICH2 including mvapich2/2.1, mvapich2/2.2 and its variants do not support Slurm very well due to its life span, so we will remove the following versions:
- mvapich2/2.1
- mvapich2/2.2, 2.2rc1, 2.2ddn1.3, 2.2ddn1.4, 2.2-debug, 2.2-gpu
As a result, the following dependent software will not be available anymore.
| Unavailable Software | Possible replacement |
|---|---|
| amber/16 | amber/18 |
| darshan/3.1.4 | darshan/3.1.6 |
| darshan/3.1.5-pre1 | darshan/3.1.6 |
| expresso/5.2.1 | expresso/6.3 |
| expresso/6.1 | expresso/6.3 |
| expresso/6.1.2 | expresso/6.3 |
| fftw3/3.3.4 | fftw3/3.3.5 |
| gamess/18Aug2016R1 | gamess/30Sep2019R2 |
| gromacs/2016.4 | gromacs/2018.2 |
| gromacs/5.1.2 | gromacs/2018.2 |
| lammps/14May16 | lammps/16Mar18 |
| lammps/31Mar17 | lammps/16Mar18 |
| mumps/5.0.2 | N/A (no current users) |
| namd/2.11 | namd/2.13 |
| nwchem/6.6 | nwchem/6.8 |
| pnetcdf/1.7.0 | pnetcdf/1.10.0 |
| siesta-par/4.0 | siesta-par/4.0.2 |
If you used one of the software listed above, we strongly recommend testing during the early user period. We listed a possible replacement version that is close to the unavailable version. However, if it is possible, we recommend using the most recent versions available. You can find the available versions by module spider {software name}. If you have any questions, please contact OSC Help.
Miscellaneous cleanup on MPIs
We clean up miscellaneous MPIs as we have a better and compatible version available. Since it has a compatible version, you should be able to use your applications without issues.
| Removed MPI versions | Compatible MPI versions |
|---|---|
|
mvapich2/2.3b mvapich2/2.3rc1 mvapich2/2.3rc2 |
mvapich2/2.3 mvapich2/2.3.3 |
|
mvapich2/2.3b-gpu mvapich2/2.3rc1-gpu mvapich2/2.3rc2-gpu mvapich2/2.3-gpu mvapich2/2.3.1-gpu mvapich2-gdr/2.3.1, 2.3.2, 2.3.3 |
mvapich2-gdr/2.3.4 |
|
openmpi/1.10.5 openmpi/1.10 |
openmpi/1.10.7 openmpi/1.10.7-hpcx |
|
openmpi/2.0 openmpi/2.0.3 openmpi/2.1.2 |
openmpi/2.1.6 openmpi/2.1.6-hpcx |
|
openmpi/4.0.2 openmpi/4.0.2-hpcx |
openmpi/4.0.3 openmpi/4.0.3-hpcx |
Software flag usage update for Licensed Software
We have software flags required to use in job scripts for licensed software, such as ansys, abauqs, or schrodinger. With the slurm migration, we updated the syntax and added extra software flags. It is very important everyone follow the procedure below. If you don't use the software flags properly, jobs submitted by others can be affected.
We require using software flags only for the demanding software and the software features in order to prevent job failures due to insufficient licenses. When you use the software flags, Slurm will record it on its license pool, so that other jobs will launch when there are enough licenses available. This will function correctly when everyone uses the software flag.
During the early user period until Dec 15, 2020, the software flag system may not work correctly. This is because, during the test period, licenses will be used from two separate Owens systems. However, we recommend you to test your job scripts with the new software flags, so that you can use it without any issues after the slurm migration.
The new syntax for software flags is
#SBATCH -L {software flag}@osc:N
where N is the requesting number of the licenses. If you need more than one software flags, you can use
#SBATCH -L {software flag1}@osc:N,{software flag2}@osc:M
For example, if you need 2 abaqus and 2 abaqusextended license features, then you can use
$SBATCH -L abaqus@osc:2,abaqusextended@osc:2
We have the full list of software associated with software flags in the table below.
| Software flag | Note | |
|---|---|---|
| abaqus |
abaqus, abaquscae |
|
| ansys | ansys, ansyspar | |
| comsol | comsolscript | |
| schrodinger | epik, glide, ligprep, macromodel, qikprop | |
| starccm | starccm, starccmpar | |
| stata | stata | |
| usearch | usearch | |
| ls-dyna, mpp-dyna | lsdyna |
Owens Programming Environment (PBS)
This document is obsoleted and kept as a reference to previous Owens programming environment. Please refer to here for the latest version.
Compilers
C, C++ and Fortran are supported on the Owens cluster. Intel, PGI and GNU compiler suites are available. The Intel development tool chain is loaded by default. Compiler commands and recommended options for serial programs are listed in the table below. See also our compilation guide.
The Haswell and Broadwell processors that make up Owens support the Advanced Vector Extensions (AVX2) instruction set, but you must set the correct compiler flags to take advantage of it. AVX2 has the potential to speed up your code by a factor of 4 or more, depending on the compiler and options you would otherwise use.
In our experience, the Intel and PGI compilers do a much better job than the GNU compilers at optimizing HPC code.
With the Intel compilers, use -xHost and -O2 or higher. With the GNU compilers, use -march=native and -O3. The PGI compilers by default use the highest available instruction set, so no additional flags are necessary.
This advice assumes that you are building and running your code on Owens. The executables will not be portable. Of course, any highly optimized builds, such as those employing the options above, should be thoroughly validated for correctness.
| LANGUAGE | INTEL EXAMPLE | PGI EXAMPLE | GNU EXAMPLE |
|---|---|---|---|
| C | icc -O2 -xHost hello.c | pgcc -fast hello.c | gcc -O3 -march=native hello.c |
| Fortran 90 | ifort -O2 -xHost hello.f90 | pgf90 -fast hello.f90 | gfortran -O3 -march=native hello.f90 |
| C++ | icpc -O2 -xHost hello.cpp | pgc++ -fast hello.cpp | g++ -O3 -march=native hello.cpp |
Parallel Programming
MPI
OSC systems use the MVAPICH2 implementation of the Message Passing Interface (MPI), optimized for the high-speed Infiniband interconnect. MPI is a standard library for performing parallel processing using a distributed-memory model. For more information on building your MPI codes, please visit the MPI Library documentation.
Parallel programs are started with the mpiexec command. For example,
mpiexec ./myprog
The mpiexec command will normally spawn one MPI process per CPU core requested in a batch job. Use the -n and/or -ppn option to change that behavior.
The table below shows some commonly used options. Use mpiexec -help for more information.
| MPIEXEC Option | COMMENT |
|---|---|
-ppn 1 |
One process per node |
-ppn procs |
procs processes per node |
-n totalprocs-np totalprocs |
At most totalprocs processes per node |
-prepend-rank |
Prepend rank to output |
-help |
Get a list of available options |
OpenMP
The Intel, PGI and GNU compilers understand the OpenMP set of directives, which support multithreaded programming. For more information on building OpenMP codes on OSC systems, please visit the OpenMP documentation.
GPU Programming
160 Nvidia P100 GPUs are available on Owens. Please visit our GPU documentation.
Owens Programming Environment
Compilers
C, C++ and Fortran are supported on the Owens cluster. Intel, PGI and GNU compiler suites are available. The Intel development tool chain is loaded by default. Compiler commands and recommended options for serial programs are listed in the table below. See also our compilation guide.
The Haswell and Broadwell processors that make up Owens support the Advanced Vector Extensions (AVX2) instruction set, but you must set the correct compiler flags to take advantage of it. AVX2 has the potential to speed up your code by a factor of 4 or more, depending on the compiler and options you would otherwise use.
In our experience, the Intel and PGI compilers do a much better job than the GNU compilers at optimizing HPC code.
With the Intel compilers, use -xHost and -O2 or higher. With the GNU compilers, use -march=native and -O3. The PGI compilers by default use the highest available instruction set, so no additional flags are necessary.
This advice assumes that you are building and running your code on Owens. The executables will not be portable. Of course, any highly optimized builds, such as those employing the options above, should be thoroughly validated for correctness.
| LANGUAGE | INTEL | GNU | PGI |
|---|---|---|---|
| C | icc -O2 -xHost hello.c | gcc -O3 -march=native hello.c | pgcc -fast hello.c |
| Fortran 77/90 | ifort -O2 -xHost hello.F | gfortran -O3 -march=native hello.F | pgfortran -fast hello.F |
| C++ | icpc -O2 -xHost hello.cpp | g++ -O3 -march=native hello.cpp | pgc++ -fast hello.cpp |
Parallel Programming
MPI
OSC systems use the MVAPICH2 implementation of the Message Passing Interface (MPI), optimized for the high-speed Infiniband interconnect. MPI is a standard library for performing parallel processing using a distributed-memory model. For more information on building your MPI codes, please visit the MPI Library documentation.
MPI programs are started with the srun command. For example,
#!/bin/bash
#SBATCH --nodes=2
srun [ options ] mpi_prog
The srun command will normally spawn one MPI process per task requested in a Slurm batch job. Use the --ntasks-per-node=n option to change that behavior. For example,
#!/bin/bash #SBATCH --nodes=2 # Use the maximum number of CPUs of two nodes srun ./mpi_prog # Run 8 processes per node srun -n 16 --ntasks-per-node=8 ./mpi_prog
The table below shows some commonly used options. Use srun -help for more information.
| OPTION | COMMENT |
|---|---|
--ntasks-per-node=n |
number of tasks to invoke on each node |
-help |
Get a list of available options |
srun in any circumstances.OpenMP
The Intel, GNU and PGI compilers understand the OpenMP set of directives, which support multithreaded programming. For more information on building OpenMP codes on OSC systems, please visit the OpenMP documentation.
An OpenMP program by default will use a number of threads equal to the number of CPUs requested in a Slurm batch job. To use a different number of threads, set the environment variable OMP_NUM_THREADS. For example,
#!/bin/bash #SBATCH --ntasks-per-node=8 # Run 8 threads ./omp_prog # Run 4 threads export OMP_NUM_THREADS=4 ./omp_prog
To run a OpenMP job on an exclusive node:
#!/bin/bash #SBATCH --nodes=1 #SBATCH --exclusive export OMP_NUM_THREADS=$SLURM_CPUS_ON_NODE ./omp_prog
Interactive job only
See the section on interactive batch in batch job submission for details on submitting an interactive job to the cluster.
Hybrid (MPI + OpenMP)
An example of running a job for hybrid code:
#!/bin/bash #SBATCH --nodes=2 # Run 4 MPI processes on each node and 7 OpenMP threads spawned from a MPI process export OMP_NUM_THREADS=7 srun -n 8 -c 7 --ntasks-per-node=4 ./hybrid_prog
Tuning Parallel Program Performance: Process/Thread Placement
To get the maximum performance, it is important to make sure that processes/threads are located as close as possible to their data, and as close as possible to each other if they need to work on the same piece of data, with given the arrangement of node, sockets, and cores, with different access to RAM and caches.
While cache and memory contention between threads/processes are an issue, it is best to use scatter distribution for code.
Processes and threads are placed differently depending on the computing resources you requste and the compiler and MPI implementation used to compile your code. For the former, see the above examples to learn how to run a job on exclusive nodes. For the latter, this section summarizes the default behavior and how to modify placement.
OpenMP only
For all three compilers (Intel, GNU, PGI), purely threaded codes do not bind to particular CPU cores by default. In other words, it is possible that multiple threads are bound to the same CPU core.
The following table describes how to modify the default placements for pure threaded code:
| DISTRIBUTION | Compact | Scatter/Cyclic |
|---|---|---|
| DESCRIPTION | Place threads as closely as possible on sockets | Distribute threads as evenly as possible across sockets |
| INTEL | KMP_AFFINITY=compact | KMP_AFFINITY=scatter |
| GNU | OMP_PLACES=sockets[1] | OMP_PROC_BIND=spread/close |
| PGI[2] |
MP_BIND=yes |
MP_BIND=yes |
- Threads in the same socket might be bound to the same CPU core.
- PGI LLVM-backend (version 19.1 and later) does not support thread/processors affinity on NUMA architecture. To enable this feature, compile threaded code with
--Mnollvmto use proprietary backend.
MPI Only
For MPI-only codes, MVAPICH2 first binds as many processes as possible on one socket, then allocates the remaining processes on the second socket so that consecutive tasks are near each other. Intel MPI and OpenMPI alternately bind processes on socket 1, socket 2, socket 1, socket 2 etc, as cyclic distribution.
For process distribution across nodes, all MPIs first bind as many processes as possible on one node, then allocates the remaining processes on the second node.
The following table describe how to modify the default placements on a single node for MPI-only code with the command srun:
| DISTRIBUTION (single node) |
Compact | Scatter/Cyclic |
|---|---|---|
| DESCRIPTION | Place processs as closely as possible on sockets | Distribute process as evenly as possible across sockets |
| MVAPICH2[1] | Default | MV2_CPU_BINDING_POLICY=scatter |
| INTEL MPI | srun --cpu-bind="map_cpu:$(seq -s, 0 2 27),$(seq -s, 1 2 27)" | Default |
| OPENMPI | srun --cpu-bind="map_cpu:$(seq -s, 0 2 27),$(seq -s, 1 2 27)" | Default |
MV2_CPU_BINDING_POLICYwill not work ifMV2_ENABLE_AFFINITY=0is set.
To distribute processes evenly across nodes, please set SLURM_DISTRIBUTION=cyclic.
Hybrid (MPI + OpenMP)
For Hybrid codes, each MPI process is allocated OMP_NUM_THREADS cores and the threads of each process are bound to those cores. All MPI processes (as well as the threads bound to the process) behave as we describe in the previous sections. It means the threads spawned from a MPI process might be bound to the same core. To change the default process/thread placmements, please refer to the tables above.
Summary
The above tables list the most commonly used settings for process/thread placement. Some compilers and Intel libraries may have additional options for process and thread placement beyond those mentioned on this page. For more information on a specific compiler/library, check the more detailed documentation for that library.
GPU Programming
160 Nvidia P100 GPUs are available on Owens. Please visit our GPU documentation.
Reference
Citation
For more information about citations of OSC, visit https://www.osc.edu/citation.
To cite Owens, please use the following Archival Resource Key:
ark:/19495/hpc6h5b1
Please adjust this citation to fit the citation style guidelines required.
Ohio Supercomputer Center. 2016. Owens Supercomputer. Columbus, OH: Ohio Supercomputer Center. http://osc.edu/ark:19495/hpc6h5b1
Here is the citation in BibTeX format:
@misc{Owens2016,
ark = {ark:/19495/hpc93fc8},
url = {http://osc.edu/ark:/19495/hpc6h5b1},
year = {2016},
author = {Ohio Supercomputer Center},
title = {Owens Supercomputer}
}
And in EndNote format:
%0 Generic %T Owens Supercomputer %A Ohio Supercomputer Center %R ark:/19495/hpc6h5b1 %U http://osc.edu/ark:/19495/hpc6h5b1 %D 2016
Here is an .ris file to better suit your needs. Please change the import option to .ris.
Owens SSH key fingerprints
These are the public key fingerprints for Owens:
owens: ssh_host_rsa_key.pub = 18:68:d4:b0:44:a8:e2:74:59:cc:c8:e3:3a:fa:a5:3f
owens: ssh_host_ed25519_key.pub = 1c:3d:f9:99:79:06:ac:6e:3a:4b:26:81:69:1a:ce:83
owens: ssh_host_ecdsa_key.pub = d6:92:d1:b0:eb:bc:18:86:0c:df:c5:48:29:71:24:af
These are the SHA256 hashes:
owens: ssh_host_rsa_key.pub = SHA256:vYIOstM2e8xp7WDy5Dua1pt/FxmMJEsHtubqEowOaxo
owens: ssh_host_ed25519_key.pub = SHA256:FSb9ZxUoj5biXhAX85tcJ/+OmTnyFenaSy5ynkRIgV8
owens: ssh_host_ecdsa_key.pub = SHA256:+fqAIqaMW/DUJDB0v/FTxMT9rkbvi/qVdMKVROHmAP4
Batch Limit Rules
Memory Limit:
A small portion of the total physical memory on each node is reserved for distributed processes. The actual physical memory available to user jobs is tabulated below.
Summary
| Node type | default and max memory per core | max memory per node |
|---|---|---|
| regular compute | 4.214 GB | 117 GB |
| huge memory | 31.104 GB | 1492 GB |
| gpu | 4.214 GB | 117 GB |
e.g. The following slurm directives will actually grant this job 3 cores, with 10 GB of memory
(since 2 cores * 4.2 GB = 8.4 GB doesn't satisfy the memory request).
#SBATCH --ntasks-per-node=2
#SBATCH --mem=10gIt is recommended to let the default memory apply unless more control over memory is needed.
Note that if an entire node is requested, then the job is automatically granted the entire node's main memory. On the other hand, if a partial node is requested, then memory is granted based on the default memory per core.
See a more detailed explanation below.
Regular Dense Compute Node
On Owens, it equates to 4,315 MB/core or 120,820 MB/node (117.98 GB/node) for the regular dense compute node.
If your job requests less than a full node ( ntasks-per-node < 28 ), it may be scheduled on a node with other running jobs. In this case, your job is entitled to a memory allocation proportional to the number of cores requested (4315 MB/core). For example, without any memory request ( mem=XXMB ), a job that requests --nodes=1 --ntasks-per-node=1 will be assigned one core and should use no more than 4315 MB of RAM, a job that requests --nodes=1 --ntasks-per-node=3 will be assigned 3 cores and should use no more than 3*4315 MB of RAM, and a job that requests --nodes=1 --ntasks-per-node=28 will be assigned the whole node (28 cores) with 118 GB of RAM.
Here is some information when you include memory request (mem=XX ) in your job. A job that requests --nodes=1 --ntasks-per-node=1 --mem=12GB will be assigned three cores and have access to 12 GB of RAM, and charged for 3 cores worth of usage (in other ways, the request --ntasks-per-node is ingored). A job that requests --nodes=1 --ntasks-per-node=5 --mem=12GB will be assigned 5 cores but have access to only 12 GB of RAM, and charged for 5 cores worth of usage.
A multi-node job ( nodes>1 ) will be assigned the entire nodes with 118 GB/node and charged for the entire nodes regardless of ppn request. For example, a job that requests --nodes=10 --ntasks-per-node=1 will be charged for 10 whole nodes (28 cores/node*10 nodes, which is 280 cores worth of usage).
Huge Memory Node
On Owens, it equates to 31,850 MB/core or 1,528,800 MB/node (1,492.96 GB/node) for a huge memory node.
To request no more than a full huge memory node, you have two options:
- The first is to specify the memory request between 120,832 MB (118 GB) and 1,528,800 MB (1,492.96 GB), i.e.,
120832MB <= mem <=1528800MB(118GB <= mem < 1493GB). Note: you can only use interger for request - The other option is to use the combination of
--ntasks-per-nodeand--partition, like--ntasks-per-node=4 --partition=hugemem. When no memory is specified for the huge memory node, your job is entitled to a memory allocation proportional to the number of cores requested (31,850MB/core). Note,--ntasks-per-nodeshould be no less than 4 and no more than 48.
To manage and monitor your memory usage, please refer to Out-of-Memory (OOM) or Excessive Memory Usage.
GPU Jobs
There is only one GPU per GPU node on Owens.
For serial jobs, we allow node sharing on GPU nodes so a job may request any number of cores (up to 28)
(--nodes=1 --ntasks-per-node=XX --gpus-per-node=1)
For parallel jobs (n>1), we do not allow node sharing.
See this GPU computing page for more information.
Partition time and job size limits
Here are the partitions available on Owens:
| Name | Max time limit (dd-hh:mm:ss) |
Min job size | Max job size | notes |
|---|---|---|---|---|
|
serial |
7-00:00:00 |
1 core |
1 node |
|
|
longserial |
14-00:00:0 |
1 core |
1 node |
|
|
parallel |
4-00:00:00 |
2 nodes |
81 nodes |
|
| gpuserial | 7-00:00:00 | 1 core | 1 node | |
| gpuparallel | 4-00:00:00 | 2 nodes | 8 nodes | |
|
hugemem |
7-00:00:00 |
1 core |
1 node |
|
| hugemem-parallel | 4-00:00:00 | 2 nodes | 16 nodes |
|
| debug | 1:00:00 | 1 core | 2 nodes |
|
| gpudebug | 1:00:00 | 1 core | 2 nodes |
|
--partition=<partition-name> to the sbatch command at submission time or add this line to the job script:#SBATCH --paritition=<partition-name>To access one of the restricted queues, please contact OSC Help. Generally, access will only be granted to these queues if the performance of the job cannot be improved, and job size cannot be reduced by splitting or checkpointing the job.
Job/Core Limits
| Max Running Job Limit | Max Core/Processor Limit | Max node Limit | ||||
|---|---|---|---|---|---|---|
| For all types | GPU jobs | Regular debug jobs | GPU debug jobs | For all types | hugemem | |
| Individual User | 384 | 132 | 4 | 4 | 3080 | 12 |
| Project/Group | 576 | 132 | n/a | n/a | 3080 | 12 |
An individual user can have up to the max concurrently running jobs and/or up to the max processors/cores in use.
However, among all the users in a particular group/project, they can have up to the max concurrently running jobs and/or up to the max processors/cores in use.
Owens cluster history by the numbers
The Owens high performance computing (HPC) cluster, decommissioned in February 2025, ran more than 36 million jobs during its time at the OSC data center. The $9.7 million system, which launched in 2016, was named for Ohio native and Olympic champion J.C. “Jesse” Owens.
At the time of its launch, Owens boasted nearly 10 times the performance power of any previous OSC system. During the cluster’s lifespan, OSC observed a growing number of clients use its GPUs for artificial intelligence and machine learning work. In addition, Owens featured a novel cooling system with rear door heat exchangers.
The cluster’s lifetime usage statistics, captured by OSC below, reflect several trends over the past decade: clients’ rising use of GPUs and Python for data analysis, as well as an increase in commercial clients.
|
Job Stats: |
Number: |
% of Total: |
|
Total jobs run |
36,154,977 |
|
|
Jobs that requested GPUs |
2,110,504 |
5.8% |
|
Jobs that requested Big Memory |
439,542 |
1.2% |
|
Jobs that used a single core |
10,197,238 |
28.2% |
|
Jobs that used no more than a single node (28 cores) |
34,916,359 |
96.6% |
|
Jobs that used more than 81 nodes / 2,268 cores (default maximum allowed) |
2,949 |
0.01% |
|
Jobs completed in less than 1 hour |
28,638,060 |
79.2% |
|
Jobs that waited less than 30 minutes in the queue |
27,777,842 |
76.8% |
Timing Stats:
-
Total core hours: 1,131,350,805
-
Total GPU hours: 6,418,404
-
Average wall time used: 1 hour, 11 minutes, 27 seconds
-
Average queue wait time: 1 hour, 3 minutes, 38 seconds
-
Longest running job: 30 days, 28 seconds
-
First job start time: 2016-08-27 12:06:07
-
Last job end time: 2025-02-03 06:46:32
-
Elapsed time between first and last jobs: 8 years, 5 months, 6 days, 15 hours, 5 minutes, 19 seconds
-
Physical delivery: 2016-06-06
-
Physical removal of last components from the State of Ohio Computer Center (SOCC): 2025-05-01
Client Stats:
-
Count of distinct projects that ran jobs: 2,292
-
Count of distinct users that ran jobs: 10,284
-
Most active user by core hours: Minkyu Kim, The Ohio State University (31,616,854 / 2.8% of total)
-
Most active user by jobs run: Balasubramanian Ganesan, Mars Incorporated (3,357,138 / 9.3% of total)
-
Most active project by core hours: PAA0023 “SIMCenter,” Ohio State (54,212,464 / 4.8% of total)
-
Most active project by jobs run: PYS1083 “Mars Genome Assembly,” Mars Incorporated (3,357,138 / 9.3% of total)
-
First non-staff member to run a job: Greg Padgett, TotalSim (2016-08-27)
-
Last non-staff member to run a job: Mason Pacenta, Ohio State (2025-02-03)
Misc Stats:
-
Most run application: Python
-
5,787,724 jobs (16.0% of total)
-
98,653,602 core hours (8.7% of total)
-
822,760 GPU hours
-
Average cores used per job: 15
-
Average lifetime utilization: 65.4%
Owens cluster transition: Action required
After eight years of service, the Owens high performance computing (HPC) cluster will be decommissioned over the next two months. Clients currently using Owens for research and classroom instruction must migrate jobs to other OSC clusters during this time.
To assist clients with transitioning their workflows, Owens will be decommissioned in two phases. On Jan. 6, 2025, OSC will move two-thirds of the regular compute nodes and one-half of the GPU nodes (a total of about 60% of the cluster cores) offline. The remainder of the nodes will power down on Feb. 3, 2025.
OSC will remove any remaining licensed software from Owens on Dec. 17, 2024. Abaqus, Ansys, LS-DYNA and Star-CCM+ are now available on the Cardinal cluster for all academic users.
Owens clients can migrate jobs to the Pitzer, Ascend or Cardinal clusters; classroom instructors should prepare all forthcoming courses on one of these systems. If you are using Jupyter/RStudio classroom apps, please prepare courses on Pitzer.
Cardinal, OSC’s newest cluster, became available for all clients to use on Nov. 4, 2024. A new ScarletCanvas course, “Getting Started with Cardinal,” provides an overview of this HPC resource and how to use it. Visit the OSC course module on ScarletCanvas to enroll in and complete the course, which is available in an asynchronous format.
In addition to launching Cardinal, OSC is transitioning the data center space occupied by the Owens cluster to accommodate an expansion of the Ascend cluster. The new resources are expected to be available for client use in March 2025. More details will be released in January.
Thank you for your cooperation in transitioning your workflows. If you discover that a software package you are using on Owens is currently not supported on the other clusters, you may install it locally or contact oschelp@osc.edu for assistance.
OSC is excited to offer our clients new and improved HPC resources that will benefit your research, classroom and innovation initiatives. For more information about our cluster transition, please visit the OSC site.