Supercomputers
We currently operate three major systems:
- Owens Cluster, a 23,000+ core Dell Intel Xeon machine
- Ruby Cluster, an 4800 core HP Intel Xeon machine
- 20 nodes have Nvidia Tesla K40 GPUs
- One node has 1 TB of RAM and 32 cores, for large SMP style jobs.
- Pitzer Cluster, an 10,500+ core Dell Intel Xeon machine
Our clusters share a common environment, and we have several guides available.
- An introduction to the basic login environment.
- A detailed guide to our batch environment, explaining how to submit jobs to the scheduler and request the required resources for your computations.
OSC also provides more than 5 PB of storage, and another 5.5 PB of tape backup.
- Learn how that space is made available to users, and how to best utilize the resources, in our storage environment guide.
Finally, you can keep up to date with any known issues on our systems (and the available workarounds). An archive of resolved issues can be found here.
Ascend
OSC's Ascend cluster was installed in fall 2022 and is a Dell-built, AMD EPYC™ CPUs with NVIDIA A100 80GB GPUs cluster devoted entirely to intensive GPU processing.
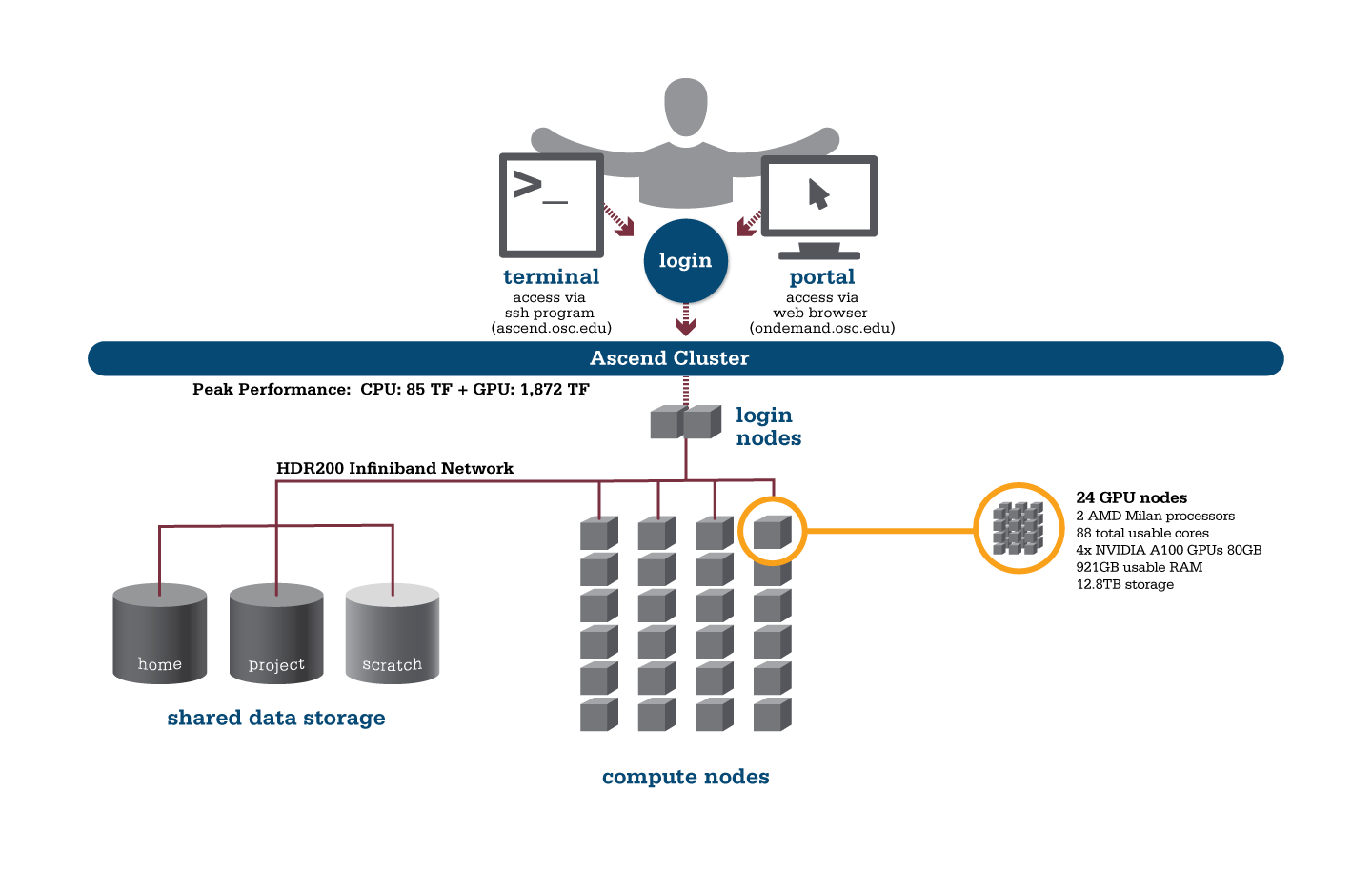
Hardware

Detailed system specifications:
- 24 Power Edge XE 8545 nodes, each with:
- 2 AMD EPYC 7643 (Milan) processors (2.3 GHz, each with 44 usable cores)
- 4 NVIDIA A100 GPUs with 80GB memory each, supercharged by NVIDIA NVLink
- 921GB usable RAM
- 12.8TB NVMe internal storage
- 2,112 total usable cores
- 88 cores/node & 921GB of memory/node
- Mellanox/NVIDA 200 Gbps HDR InfiniBand
- Theoretical system peak performance
- 1.95 petaflops
- 2 login nodes
- IP address: 192.148.247.[180-181]
How to Connect
-
SSH Method
To login to Ascend at OSC, ssh to the following hostname:
ascend.osc.edu
You can either use an ssh client application or execute ssh on the command line in a terminal window as follows:
ssh <username>@ascend.osc.edu
You may see a warning message including SSH key fingerprint. Verify that the fingerprint in the message matches one of the SSH key fingerprints listed here, then type yes.
From there, you are connected to the Ascend login node and have access to the compilers and other software development tools. You can run programs interactively or through batch requests. We use control groups on login nodes to keep the login nodes stable. Please use batch jobs for any compute-intensive or memory-intensive work. See the following sections for details.
-
OnDemand Method
You can also login to Ascend at OSC with our OnDemand tool. The first step is to log into OnDemand. Then once logged in you can access Ascend by clicking on "Clusters", and then selecting ">_Ascend Shell Access".
Instructions on how to connect to OnDemand can be found at the OnDemand documentation page.
File Systems
Ascend accesses the same OSC mass storage environment as our other clusters. Therefore, users have the same home directory as on the old clusters. Full details of the storage environment are available in our storage environment guide.
Software Environment
The module system on Ascend is the same as on the Owens and Pitzer systems. Use module load <package> to add a software package to your environment. Use module list to see what modules are currently loaded and module avail to see the modules that are available to load. To search for modules that may not be visible due to dependencies or conflicts, use module spider .
You can keep up to the software packages that have been made available on Ascend by viewing the Software by System page and selecting the Ascend system.
Batch Specifics
Refer to this Slurm migration page to understand how to use Slurm on the Ascend cluster.
Using OSC Resources
For more information about how to use OSC resources, please see our guide on batch processing at OSC. For specific information about modules and file storage, please see the Batch Execution Environment page.
Ascend Programming Environment
Compilers
C, C++ and Fortran are supported on the Ascend cluster. Intel, oneAPI, GNU, nvhpc, and aocc compiler suites are available. The Intel development tool chain is loaded by default. Compiler commands and recommended options for serial programs are listed in the table below. See also our compilation guide.
The Milan processors from AMD that make up Ascend support the Advanced Vector Extensions (AVX2) instruction set, but you must set the correct compiler flags to take advantage of it. AVX2 has the potential to speed up your code by a factor of 4 or more, depending on the compiler and options you would otherwise use. However, bare in mind that clock speeds decrease as the level of the instruction set increases. So, if your code does not benefit from vectorization it may be beneficial to use a lower instruction set.
In our experience, the Intel compiler usually does the best job of optimizing numerical codes and we recommend that you give it a try if you’ve been using another compiler.
With the Intel compilers, use -xHost and -O2 or higher. With the GNU compilers, use -march=native and -O3.
This advice assumes that you are building and running your code on Ascend. The executables will not be portable. Of course, any highly optimized builds, such as those employing the options above, should be thoroughly validated for correctness.
| LANGUAGE | INTEL | GNU |
|---|---|---|
| C | icc -O2 -xHost hello.c | gcc -O3 -march=native hello.c |
| Fortran 77/90 | ifort -O2 -xHost hello.F | gfortran -O3 -march=native hello.F |
| C++ | icpc -O2 -xHost hello.cpp | g++ -O3 -march=native hello.cpp |
Parallel Programming
MPI
OSC systems use the MVAPICH2 implementation of the Message Passing Interface (MPI), optimized for the high-speed Infiniband interconnect. MPI is a standard library for performing parallel processing using a distributed-memory model. For more information on building your MPI codes, please visit the MPI Library documentation.
MPI programs are started with the srun command. For example,
#!/bin/bash
#SBATCH --nodes=2
#SBATCH --ntasks-per-node=84
srun [ options ] mpi_prog
The srun command will normally spawn one MPI process per task requested in a Slurm batch job. Use the -n ntasks and/or --ntasks-per-node=n option to change that behavior. For example,
#!/bin/bash #SBATCH --nodes=2 # Use the maximum number of CPUs of two nodes srun ./mpi_prog # Run 8 processes per node srun -n 16 --ntasks-per-node=8 ./mpi_prog
The table below shows some commonly used options. Use srun -help for more information.
| OPTION | COMMENT |
|---|---|
-n, --ntasks=ntasks |
total number of tasks to run |
--ntasks-per-node=n |
number of tasks to invoke on each node |
-help |
Get a list of available options |
srun in any circumstances.OpenMP
The Intel, and GNU compilers understand the OpenMP set of directives, which support multithreaded programming. For more information on building OpenMP codes on OSC systems, please visit the OpenMP documentation.
An OpenMP program by default will use a number of threads equal to the number of CPUs requested in a Slurm batch job. To use a different number of threads, set the environment variable OMP_NUM_THREADS. For example,
#!/bin/bash #SBATCH --ntasks=8 # Run 8 threads ./omp_prog # Run 4 threads export OMP_NUM_THREADS=4 ./omp_prog
Interactive job only
Please use -c, --cpus-per-task=X instead of -n, --ntasks=X to request an interactive job. Both result in an interactive job with X CPUs available but only the former option automatically assigns the correct number of threads to the OpenMP program. If the option --ntasks is used only, the OpenMP program will use one thread or all threads will be bound to one CPU core.
Hybrid (MPI + OpenMP)
An example of running a job for hybrid code:
#!/bin/bash #SBATCH --nodes=2 #SBATCH --ntasks-per-node=80 # Run 4 MPI processes on each node and 40 OpenMP threads spawned from a MPI process export OMP_NUM_THREADS=40 srun -n 8 -c 40 --ntasks-per-node=4 ./hybrid_prog
Tuning Parallel Program Performance: Process/Thread Placement
To get the maximum performance, it is important to make sure that processes/threads are located as close as possible to their data, and as close as possible to each other if they need to work on the same piece of data, with given the arrangement of node, sockets, and cores, with different access to RAM and caches.
While cache and memory contention between threads/processes are an issue, it is best to use scatter distribution for code.
Processes and threads are placed differently depending on the computing resources you requste and the compiler and MPI implementation used to compile your code. For the former, see the above examples to learn how to run a job on exclusive nodes. For the latter, this section summarizes the default behavior and how to modify placement.
OpenMP only
For all two compilers (Intel, GNU), purely threaded codes do not bind to particular CPU cores by default. In other words, it is possible that multiple threads are bound to the same CPU core.
The following table describes how to modify the default placements for pure threaded code:
| DISTRIBUTION | Compact | Scatter/Cyclic |
|---|---|---|
| DESCRIPTION | Place threads close to each other as possible in successive order | Distribute threads as evenly as possible across sockets |
| INTEL | KMP_AFFINITY=compact | KMP_AFFINITY=scatter |
| GNU | OMP_PROC_BIND=true OMP_PLACE=cores |
OMP_PROC_BIND=true OMP_PLACE="{0},{48},{1},{49},..."[1] |
- The core IDs on the first and second sockets start with 0 and 48, respectively.
MPI Only
For MPI-only codes, MVAPICH2 first binds as many processes as possible on one socket, then allocates the remaining processes on the second socket so that consecutive tasks are near each other. Intel MPI and OpenMPI alternately bind processes on socket 1, socket 2, socket 1, socket 2 etc, as cyclic distribution.
For process distribution across nodes, all MPIs first bind as many processes as possible on one node, then allocates the remaining processes on the second node.
The following table describe how to modify the default placements on single node for MPI-only code with the command srun:
| DISTRIBUTION (single node) |
Compact | Scatter/Cyclic |
|---|---|---|
| DESCRIPTION | Place processs close to each other as possible in successive order | Distribute process as evenly as possible across sockets |
| MVAPICH2[1] | Default | MV2_CPU_BINDING_POLICY=scatter |
| INTEL MPI | srun --ntasks=84 --cpu-bind="map_cpu:$(seq -s, 0 43),$(seq -s, 48 95)" | Default |
| OPENMPI | srun --ntasks=84 --cpu-bind="map_cpu:$(seq -s, 0 43),$(seq -s, 48 95)" | Default |
MV2_CPU_BINDING_POLICYwill not work ifMV2_ENABLE_AFFINITY=0is set.
To distribute processes evenly across nodes, please set SLURM_DISTRIBUTION=cyclic.
Hybrid (MPI + OpenMP)
For Hybrid codes, each MPI process is allocated OMP_NUM_THREADS cores and the threads of each process are bound to those cores. All MPI processes (as well as the threads bound to the process) behave as we describe in the previous sections. It means the threads spawned from a MPI process might be bound to the same core. To change the default process/thread placmements, please refer to the tables above.
Summary
The above tables list the most commonly used settings for process/thread placement. Some compilers and Intel libraries may have additional options for process and thread placement beyond those mentioned on this page. For more information on a specific compiler/library, check the more detailed documentation for that library.
GPU Programming
96 NVIDIA A100 GPUs are available on Ascend. Please visit our GPU documentation.
Reference
Batch Limit Rules
Memory limit
It is strongly suggested to consider the memory use to the available per-core memory when users request OSC resources for their jobs.
Summary
| Node type | default memory per core (GB) | max usable memory per node (GB) |
|---|---|---|
| gpu (4 gpus) - 88 cores | 10.4726 GB | 921.5937 GB |
It is recommended to let the default memory apply unless more control over memory is needed.
Note that if an entire node is requested, then the job is automatically granted the entire node's main memory. On the other hand, if a partial node is requested, then memory is granted based on the default memory per core.
See a more detailed explanation below.
Default memory limits
A job can request resources and allow the default memory to apply. If a job requires 300 GB for example:
#SBATCH --ntasks=1 #SBATCH --cpus-per-task=30
This requests 30 cores, and each core will automatically be allocated 10.4 GB of memory (30 core * 10 GB memory = 300 GB memory).
Explicit memory requests
If needed, an explicit memory request can be added:
#SBATCH --ntasks=1 #SBATCH --cpus-per-task=4 #SBATCH --mem=300G
See Job and storage charging for details.
CPU only jobs
Dense gpu nodes on Ascend have 88 cores each. However, cpuonly partition jobs may only request 84 cores per node.
An example request would look like:
#!/bin/bash #SBATCH --partition=cpuonly #SBATCH --time=5:00:00 #SBATCH --nodes=1 #SBATCH --ntasks=2 #SBATCH --cpus-per-task=42 # requests 2 tasks * 42 cores each = 84 cores <snip>
GPU Jobs
Jobs may request only parts of gpu node. These jobs may request up to the total cores on the node (88 cores).
Requests two gpus for one task:
#SBATCH --time=5:00:00 #SBATCH --nodes=1 #SBATCH --ntasks=1 #SBATCH --cpus-per-task=20 #SBATCH --gpus-per-task=2
Requests two gpus, one for each task:
#SBATCH --time=5:00:00 #SBATCH --nodes=1 #SBATCH --ntasks=2 #SBATCH --cpus-per-task=10 #SBATCH --gpus-per-task=1
Of course, jobs can request all the gpus of a dense gpu node as well. These jobs have access to all cores as well.
Request an entire dense gpu node:
#SBATCH --nodes=1 #SBATCH --ntasks=1 #SBATCH --cpus-per-task=88 #SBATCH --gpus-per-node=4
Partition time and job size limits
Here is the walltime and node limits per job for different queues/partitions available on Ascend:
|
NAME |
MAX TIME LIMIT |
MIN JOB SIZE |
MAX JOB SIZE |
NOTES |
|---|---|---|---|---|
|
cpuonly |
4-00:00:00 |
1 core |
4 nodes |
This partition may not request gpus 84 cores per node only |
| gpu |
7-00:00:00 |
1 core |
4 nodes |
|
| debug | 1:00:00 | 1 core | 2 nodes |
Usually, you do not need to specify the partition for a job and the scheduler will assign the right partition based on the requested resources. To specify a partition for a job, either add the flag --partition=<partition-name> to the sbatch command at submission time or add this line to the job script:#SBATCH --paritition=<partition-name>
Job/Core Limits
| Max Running Job Limit | Max Core/Processor Limit | Max GPU limit | ||
|---|---|---|---|---|
| For all types | GPU debug jobs | For all types | ||
| Individual User | 256 | 4 |
704 |
32 |
| Project/Group | 512 | n/a | 704 | 32 |
An individual user can have up to the max concurrently running jobs and/or up to the max processors/cores in use. However, among all the users in a particular group/project, they can have up to the max concurrently running jobs and/or up to the max processors/cores in use.
Migrating jobs from other clusters
This page includes a summary of differences to keep in mind when migrating jobs from other clusters to Ascend.
Guidance for Pitzer Users
Hardware Specifications
| Ascend (PER NODE) | Pitzer (PER NODE) | ||
|---|---|---|---|
| Regular compute node | n/a |
40 cores and 192GB of RAM 48 cores and 192GB of RAM |
|
| Huge memory node |
n/a |
48 cores and 768GB of RAM (12 nodes in this class) 80 cores and 3.0 TB of RAM (4 nodes in this class) |
|
| GPU Node |
88 cores and 921GB RAM 4 GPUs per node (24 nodes in this class) |
40 cores and 192GB of RAM, 2 GPUs per node (32 nodes in this class) 48 cores and 192GB of RAM, 2 GPUs per node (42 nodes in this class) |
|
Guidance for Owens Users
Hardware Specifications
| Ascend (PER NODE) | Owens (PER NODE) | ||
|---|---|---|---|
| Regular compute node | n/a | 28 cores and 125GB of RAM | |
| Huge memory node | n/a |
48 cores and 1.5TB of RAM (16 nodes in this class) |
|
| GPU node |
88 cores and 921GB RAM 4 GPUs per node (24 nodes in this class) |
28 cores and 125GB of RAM, 1 GPU per node (160 nodes in this class) |
|
File Systems
Ascend accesses the same OSC mass storage environment as our other clusters. Therefore, users have the same home directory, project space, and scratch space as on the other clusters.
Software Environment
Ascend uses the same module system as other OSC Clusters.
Use module load <package> to add a software package to your environment. Use module list to see what modules are currently loaded and module avail to see the modules that are available to load. To search for modules that may not be visible due to dependencies or conflicts, use module spider .
You can keep up to on the software packages that have been made available on Ascend by viewing the Software by System page and selecting the Ascend system.
Programming Environment
C, C++ and Fortran are supported on the Ascend cluster. Intel, oneAPI, GNU, nvhpc, and aocc compiler suites are available. The Intel development tool chain is loaded by default. To switch to a different compiler, use module swap . Ascend also uses the MVAPICH2 implementation of the Message Passing Interface (MPI).
See the Ascend Programming Environment page for details.
Ascend SSH key fingerprints
These are the public key fingerprints for Ascend:
ascend: ssh_host_rsa_key.pub = 2f:ad:ee:99:5a:f4:7f:0d:58:8f:d1:70:9d:e4:f4:16
ascend: ssh_host_ed25519_key.pub = 6b:0e:f1:fb:10:da:8c:0b:36:12:04:57:2b:2c:2b:4d
ascend: ssh_host_ecdsa_key.pub = f4:6f:b5:d2:fa:96:02:73:9a:40:5e:cf:ad:6d:19:e5
These are the SHA256 hashes:
ascend: ssh_host_rsa_key.pub = SHA256:4l25PJOI9sDUaz9NjUJ9z/GIiw0QV/h86DOoudzk4oQ
ascend: ssh_host_ed25519_key.pub = SHA256:pvz/XrtS+PPv4nsn6G10Nfc7yM7CtWoTnkgQwz+WmNY
ascend: ssh_host_ecdsa_key.pub = SHA256:giMUelxDSD8BTWwyECO10SCohi3ahLPBtkL2qJ3l080
Citation
For more information about citations of OSC, visit https://www.osc.edu/citation.
To cite Ascend, please use the following Archival Resource Key:
ark:/19495/hpc3ww9d
Please adjust this citation to fit the citation style guidelines required.
Ohio Supercomputer Center. 2022. Ascend Supercomputer. Columbus, OH: Ohio Supercomputer Center. http://osc.edu/ark:/19495/hpc3ww9d
Here is the citation in BibTeX format:
@misc{Ascend2022,
ark = {ark:/19495/hpc3ww9d},
url = {http://osc.edu/ark:/19495/hpc3ww9d},
year = {2022},
author = {Ohio Supercomputer Center},
title = {Ascend Supercomputer}
}
And in EndNote format:
%0 Generic %T Ascend Supercomputer %A Ohio Supercomputer Center %R ark:/19495/hpc3ww9d %U http://osc.edu/ark:/19495/hpc3ww9d %D 2022
Request access
Users who would like to use the Ascend cluster will need to request access. This is because of the particulars of the Ascend environment, which includes its size, GPUs, and scheduling policies.
Motivation
Access to Ascend is done on a case by case basis because:
- All nodes on Ascend are with 4 GPUs, and therefore it favors GPU work instead of CPU-only work
- It is a smaller machine than Pitzer and Owens, and thus has limited space for users
Good Ascend Workload Characteristics
Those interested in using Ascend should check that their work is well suited for it by using the following list. Ideal workloads will exhibit one or more of the following characteristics:
- Needs access to Ascend specific hardware (GPUs, or AMD)
- Software:
- Supports GPUs
- Takes advantage of:
- Long vector length
- Higher core count
- Improved memory bandwidth
Applying for Access
PIs of groups that would like to be considered for Ascend access should send the following in a email to OSC Help:
- Name
- Username
- Project code (group)
- Software/packages used on Ascend
- Evidence of workload being well suited for Ascend
Technical Specifications
The following are technical specifications for Ascend.
- Number of Nodes
-
24 nodes
- Number of CPU Sockets
-
48 (2 sockets/node)
- Number of CPU Cores
-
2,304 (96 cores/node)
- Cores Per Node
-
96 cores/node (88 usable cores/node)
- Internal Storage
-
12.8 TB NVMe internal storage
- Compute CPU Specifications
-
AMD EPYC 7643 (Milan) processors for compute
- 2.3 GHz
- 48 cores per processor
- Computer Server Specifications
-
24 Dell XE8545 servers
- Accelerator Specifications
-
4 NVIDIA A100 GPUs with 80GB memory each, supercharged by NVIDIA NVLink
- Number of Accelerator Nodes
-
24 total
- Total Memory
- ~ 24 TB
- Physical Memory Per Node
-
1 TB
- Physical Memory Per Core
-
10.6 GB
- Interconnect
-
Mellanox/NVIDA 200 Gbps HDR InfiniBand
Cardinal
Detailed system specifications:
-
378 Dell Nodes, 39,312 total cores, 128 GPUs
-
Dense Compute: 326 Dell PowerEdge C6620 two-socket servers, each with:
-
2 Intel Xeon CPU Max 9470 (Sapphire Rapids, 52 cores [48 usable], 2.0 GHz) processors
-
128 GB HBM2e and 512 GB DDR5 memory
-
1.6 TB NVMe local storage
-
NDR200 Infiniband
-
-
GPU Compute: 32 Dell PowerEdge XE9640 two-socket servers, each with:
-
2 Intel Xeon Platinum 8470 (Sapphire Rapids, 52 cores [48 usable], 2.0 GHz) processors
-
1 TB DDR5 memory
-
4 NVIDIA H100 (Hopper) GPUs each with 94 GB HBM2e memory and NVIDIA NVLink
-
12.8 TB NVMe local storage
-
Four NDR400 Infiniband HCAs supporting GPUDirect
-
-
Analytics: 16 Dell PowerEdge R660 two-socket servers, each with:
-
2 Intel Xeon CPU Max 9470 (Sapphire Rapids, 52 cores [48 usable], 2.0 GHz) processors
-
128 GB HBM2e and 2 TB DDR5 memory
-
12.8 TB NVMe local storage
-
NDR200 Infiniband
-
-
Login nodes: 4 Dell PowerEdge R660 two-socket servers, each with:
-
2 Intel Xeon CPU Max 9470 (Sapphire Rapids, 52 cores [48 usable], 2.0 GHz) processors
-
128 GB HBM and 1 TB DDR5 memory
-
3.2 TB NVMe local storage
-
NDR200 Infiniband
-
IP address: TBD
-
-
~10.5 PF Theoretical system peak performance
-
~8 PetaFLOPs (GPU)
-
~2.5 PetaFLOPS (CPU)
-
-
9 Physical racks, plus Two Coolant Distribution Units (CDUs) providing direct-to-the-chip liquid cooling for all nodes
Technical Specifications
The following are technical specifications for Cardinal.
- Number of Nodes
-
378 nodes
- Number of CPU Sockets
-
756 (2 sockets/node for all nodes)
- Number of CPU Cores
-
39,312
- Cores Per Node
-
104 cores/node for all nodes (96 usable)
- Local Disk Space Per Node
-
- 1.6 TB for compute nodes
- 12.8 TB for GPU and Large mem nodes
- 3.2 TB for login nodes
- Compute, Large Mem & Login Node CPU Specifications
- Intel Xeon CPU Max 9470 HBM2e (Sapphire Rapids)
- 2.0 GHz
- 52 cores per processor (48 usable)
- GPU Node CPU Specifications
- Intel Xeon Platinum 8470 (Sapphire Rapids)
- 2.0 GHz
- 52 cores per processor
- Server Specifications
-
- 326 Dell PowerEdge C6620
- 32 Dell PowerEdge XE9640 (GPU nodes)
- 20 Dell PowerEdge R660 (largemem & login nodes)
- Accelerator Specifications
-
NVIDIA H100 (Hopper) GPUs each with 96 GB HBM2e memory and NVIDIA NVLINK
- Number of Accelerator Nodes
-
32 quad GPU nodes (4 GPUs per node)
- Total Memory
-
~281 TB (44 TB HBM, 237 TB DDR5)
- Memory Per Node
-
- 128 GB HBM / 512 GB DDR5 (compute nodes)
- 1 TB (GPU nodes)
- 128 GB HBM / 2 TB DDR5 (large mem nodes)
- 128 GB HBM / 1 TB DDR5 (login nodes)
- Memory Per Core
-
- 1.2 GB HBM / 4.9 GB DDR5 (compute nodes)
- 9.8 GB (GPU nodes)
- 1.2 GB HBM / 19.7 GB DDR5 (large mem nodes)
- 1.2 GB HBM / 9.8 GB DDR5 (login nodes)
- Interconnect
-
- NDR200 Infiniband (200 Gbps) (compute, large mem, login nodes)
- 4x NDR400 Infiniband (400 Gbps x 4) with GPUDirect, allowing non-blocking communication between up to 10 nodes (GPU nodes)
Owens
OSC's Owens cluster being installed in 2016 is a Dell-built, Intel® Xeon® processor-based supercomputer.
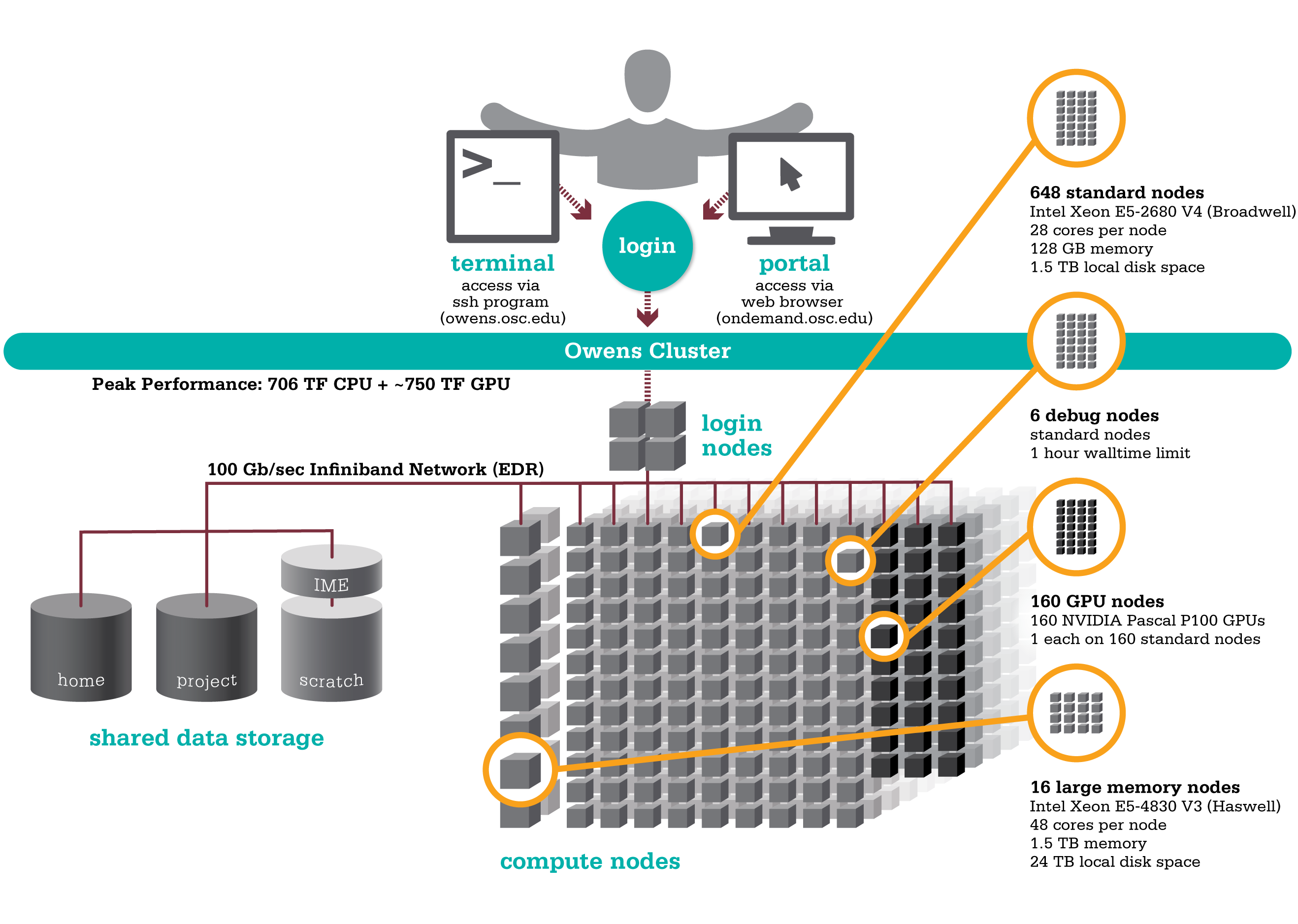 ,
,Hardware

Detailed system specifications:
Please see owens batch limits for details on user available memory amount.
- 824 Dell Nodes
- Dense Compute
-
648 compute nodes (Dell PowerEdge C6320 two-socket servers with Intel Xeon E5-2680 v4 (Broadwell, 14 cores, 2.40 GHz) processors, 128 GB memory)
-
-
GPU Compute
-
160 ‘GPU ready’ compute nodes -- Dell PowerEdge R730 two-socket servers with Intel Xeon E5-2680 v4 (Broadwell, 14 cores, 2.40 GHz) processors, 128 GB memory
-
NVIDIA Tesla P100 (Pascal) GPUs -- 5.3 TF peak (double precision), 16 GB memory
-
-
Analytics
-
16 huge memory nodes (Dell PowerEdge R930 four-socket server with Intel Xeon E5-4830 v3 (Haswell 12 core, 2.10 GHz) processors, 1,536 GB memory, 12 x 2 TB drives)
-
- 23,392 total cores
- 28 cores/node & 128 GB of memory/node
- Mellanox EDR (100 Gbps) Infiniband networking
- Theoretical system peak performance
- ~750 teraflops (CPU only)
- 4 login nodes:
- Intel Xeon E5-2680 (Broadwell) CPUs
- 28 cores/node and 256 GB of memory/node
- IP address: 192.148.247.[141-144]
How to Connect
-
SSH Method
To login to Owens at OSC, ssh to the following hostname:
owens.osc.edu
You can either use an ssh client application or execute ssh on the command line in a terminal window as follows:
ssh <username>@owens.osc.edu
You may see warning message including SSH key fingerprint. Verify that the fingerprint in the message matches one of the SSH key fingerprint listed here, then type yes.
From there, you are connected to Owens login node and have access to the compilers and other software development tools. You can run programs interactively or through batch requests. We use control groups on login nodes to keep the login nodes stable. Please use batch jobs for any compute-intensive or memory-intensive work. See the following sections for details.
-
OnDemand Method
You can also login to Owens at OSC with our OnDemand tool. The first step is to login to OnDemand. Then once logged in you can access Owens by clicking on "Clusters", and then selecting ">_Owens Shell Access".
Instructions on how to connect to OnDemand can be found at the OnDemand documention page.
File Systems
Owens accesses the same OSC mass storage environment as our other clusters. Therefore, users have the same home directory as on the old clusters. Full details of the storage environment are available in our storage environment guide.
Software Environment
The module system is used to manage the software environment on owens. Use module load <package> to add a software package to your environment. Use module list to see what modules are currently loaded and module avail to see the modules that are available to load. To search for modules that may not be visible due to dependencies or conflicts, use module spider . By default, you will have the batch scheduling software modules, the Intel compiler and an appropriate version of mvapich2 loaded.
You can keep up to on the software packages that have been made available on Owens by viewing the Software by System page and selecting the Owens system.
Compiling Code to Use Advanced Vector Extensions (AVX2)
The Haswell and Broadwell processors that make up Owens support the Advanced Vector Extensions (AVX2) instruction set, but you must set the correct compiler flags to take advantage of it. AVX2 has the potential to speed up your code by a factor of 4 or more, depending on the compiler and options you would otherwise use.
In our experience, the Intel and PGI compilers do a much better job than the gnu compilers at optimizing HPC code.
With the Intel compilers, use -xHost and -O2 or higher. With the gnu compilers, use -march=native and -O3 . The PGI compilers by default use the highest available instruction set, so no additional flags are necessary.
This advice assumes that you are building and running your code on Owens. The executables will not be portable. Of course, any highly optimized builds, such as those employing the options above, should be thoroughly validated for correctness.
See the Owens Programming Environment page for details.
Batch Specifics
Refer to the documentation for our batch environment to understand how to use the batch system on OSC hardware. Some specifics you will need to know to create well-formed batch scripts:
- Most compute nodes on Owens have 28 cores/processors per node. Huge-memory (analytics) nodes have 48 cores/processors per node.
- Jobs on Owens may request partial nodes.
Using OSC Resources
For more information about how to use OSC resources, please see our guide on batch processing at OSC. For specific information about modules and file storage, please see the Batch Execution Environment page.
Technical Specifications
The following are technical specifications for Owens.
- Number of Nodes
-
824 nodes
- Number of CPU Sockets
-
1,648 (2 sockets/node)
- Number of CPU Cores
-
23,392 (28 cores/node)
- Cores Per Node
-
28 cores/node (48 cores/node for Huge Mem Nodes)
- Local Disk Space Per Node
-
~1,500GB in /tmp
- Compute CPU Specifications
-
Intel Xeon E5-2680 v4 (Broadwell) for compute
- 2.4 GHz
- 14 cores per processor
- Computer Server Specifications
-
- 648 Dell PowerEdge C6320
- 160 Dell PowerEdge R730 (for accelerator nodes)
- Accelerator Specifications
-
NVIDIA P100 "Pascal" GPUs 16GB memory
- Number of Accelerator Nodes
-
160 total
- Total Memory
- ~ 127 TB
- Memory Per Node
-
128 GB (1.5 TB for Huge Mem Nodes)
- Memory Per Core
-
4.5 GB (31 GB for Huge Mem)
- Interconnect
-
Mellanox EDR Infiniband Networking (100Gbps)
- Login Specifications
-
4 Intel Xeon E5-2680 (Broadwell) CPUs
- 28 cores/node and 256GB of memory/node
- Special Nodes
-
16 Huge Memory Nodes
- Dell PowerEdge R930
- 4 Intel Xeon E5-4830 v3 (Haswell)
- 12 Cores
- 2.1 GHz
- 48 cores (12 cores/CPU)
- 1.5 TB Memory
- 12 x 2 TB Drive (20TB usable)
Environment changes in Slurm migration
As we migrate to Slurm from Torque/Moab, there will be necessary software environment changes.
Decommissioning old MVAPICH2 versions
Old MVAPICH2 including mvapich2/2.1, mvapich2/2.2 and its variants do not support Slurm very well due to its life span, so we will remove the following versions:
- mvapich2/2.1
- mvapich2/2.2, 2.2rc1, 2.2ddn1.3, 2.2ddn1.4, 2.2-debug, 2.2-gpu
As a result, the following dependent software will not be available anymore.
| Unavailable Software | Possible replacement |
|---|---|
| amber/16 | amber/18 |
| darshan/3.1.4 | darshan/3.1.6 |
| darshan/3.1.5-pre1 | darshan/3.1.6 |
| expresso/5.2.1 | expresso/6.3 |
| expresso/6.1 | expresso/6.3 |
| expresso/6.1.2 | expresso/6.3 |
| fftw3/3.3.4 | fftw3/3.3.5 |
| gamess/18Aug2016R1 | gamess/30Sep2019R2 |
| gromacs/2016.4 | gromacs/2018.2 |
| gromacs/5.1.2 | gromacs/2018.2 |
| lammps/14May16 | lammps/16Mar18 |
| lammps/31Mar17 | lammps/16Mar18 |
| mumps/5.0.2 | N/A (no current users) |
| namd/2.11 | namd/2.13 |
| nwchem/6.6 | nwchem/6.8 |
| pnetcdf/1.7.0 | pnetcdf/1.10.0 |
| siesta-par/4.0 | siesta-par/4.0.2 |
If you used one of the software listed above, we strongly recommend testing during the early user period. We listed a possible replacement version that is close to the unavailable version. However, if it is possible, we recommend using the most recent versions available. You can find the available versions by module spider {software name}. If you have any questions, please contact OSC Help.
Miscellaneous cleanup on MPIs
We clean up miscellaneous MPIs as we have a better and compatible version available. Since it has a compatible version, you should be able to use your applications without issues.
| Removed MPI versions | Compatible MPI versions |
|---|---|
|
mvapich2/2.3b mvapich2/2.3rc1 mvapich2/2.3rc2 |
mvapich2/2.3 mvapich2/2.3.3 |
|
mvapich2/2.3b-gpu mvapich2/2.3rc1-gpu mvapich2/2.3rc2-gpu mvapich2/2.3-gpu mvapich2/2.3.1-gpu mvapich2-gdr/2.3.1, 2.3.2, 2.3.3 |
mvapich2-gdr/2.3.4 |
|
openmpi/1.10.5 openmpi/1.10 |
openmpi/1.10.7 openmpi/1.10.7-hpcx |
|
openmpi/2.0 openmpi/2.0.3 openmpi/2.1.2 |
openmpi/2.1.6 openmpi/2.1.6-hpcx |
|
openmpi/4.0.2 openmpi/4.0.2-hpcx |
openmpi/4.0.3 openmpi/4.0.3-hpcx |
Software flag usage update for Licensed Software
We have software flags required to use in job scripts for licensed software, such as ansys, abauqs, or schrodinger. With the slurm migration, we updated the syntax and added extra software flags. It is very important everyone follow the procedure below. If you don't use the software flags properly, jobs submitted by others can be affected.
We require using software flags only for the demanding software and the software features in order to prevent job failures due to insufficient licenses. When you use the software flags, Slurm will record it on its license pool, so that other jobs will launch when there are enough licenses available. This will function correctly when everyone uses the software flag.
During the early user period until Dec 15, 2020, the software flag system may not work correctly. This is because, during the test period, licenses will be used from two separate Owens systems. However, we recommend you to test your job scripts with the new software flags, so that you can use it without any issues after the slurm migration.
The new syntax for software flags is
#SBATCH -L {software flag}@osc:N
where N is the requesting number of the licenses. If you need more than one software flags, you can use
#SBATCH -L {software flag1}@osc:N,{software flag2}@osc:M
For example, if you need 2 abaqus and 2 abaqusextended license features, then you can use
$SBATCH -L abaqus@osc:2,abaqusextended@osc:2
We have the full list of software associated with software flags in the table below.
| Software flag | Note | |
|---|---|---|
| abaqus |
abaqus, abaquscae |
|
| ansys | ansys, ansyspar | |
| comsol | comsolscript | |
| schrodinger | epik, glide, ligprep, macromodel, qikprop | |
| starccm | starccm, starccmpar | |
| stata | stata | |
| usearch | usearch | |
| ls-dyna, mpp-dyna | lsdyna |
Owens Programming Environment (PBS)
This document is obsoleted and kept as a reference to previous Owens programming environment. Please refer to here for the latest version.
Compilers
C, C++ and Fortran are supported on the Owens cluster. Intel, PGI and GNU compiler suites are available. The Intel development tool chain is loaded by default. Compiler commands and recommended options for serial programs are listed in the table below. See also our compilation guide.
The Haswell and Broadwell processors that make up Owens support the Advanced Vector Extensions (AVX2) instruction set, but you must set the correct compiler flags to take advantage of it. AVX2 has the potential to speed up your code by a factor of 4 or more, depending on the compiler and options you would otherwise use.
In our experience, the Intel and PGI compilers do a much better job than the GNU compilers at optimizing HPC code.
With the Intel compilers, use -xHost and -O2 or higher. With the GNU compilers, use -march=native and -O3. The PGI compilers by default use the highest available instruction set, so no additional flags are necessary.
This advice assumes that you are building and running your code on Owens. The executables will not be portable. Of course, any highly optimized builds, such as those employing the options above, should be thoroughly validated for correctness.
| LANGUAGE | INTEL EXAMPLE | PGI EXAMPLE | GNU EXAMPLE |
|---|---|---|---|
| C | icc -O2 -xHost hello.c | pgcc -fast hello.c | gcc -O3 -march=native hello.c |
| Fortran 90 | ifort -O2 -xHost hello.f90 | pgf90 -fast hello.f90 | gfortran -O3 -march=native hello.f90 |
| C++ | icpc -O2 -xHost hello.cpp | pgc++ -fast hello.cpp | g++ -O3 -march=native hello.cpp |
Parallel Programming
MPI
OSC systems use the MVAPICH2 implementation of the Message Passing Interface (MPI), optimized for the high-speed Infiniband interconnect. MPI is a standard library for performing parallel processing using a distributed-memory model. For more information on building your MPI codes, please visit the MPI Library documentation.
Parallel programs are started with the mpiexec command. For example,
mpiexec ./myprog
The mpiexec command will normally spawn one MPI process per CPU core requested in a batch job. Use the -n and/or -ppn option to change that behavior.
The table below shows some commonly used options. Use mpiexec -help for more information.
| MPIEXEC Option | COMMENT |
|---|---|
-ppn 1 |
One process per node |
-ppn procs |
procs processes per node |
-n totalprocs-np totalprocs |
At most totalprocs processes per node |
-prepend-rank |
Prepend rank to output |
-help |
Get a list of available options |
OpenMP
The Intel, PGI and GNU compilers understand the OpenMP set of directives, which support multithreaded programming. For more information on building OpenMP codes on OSC systems, please visit the OpenMP documentation.
GPU Programming
160 Nvidia P100 GPUs are available on Owens. Please visit our GPU documentation.
Owens Programming Environment
Compilers
C, C++ and Fortran are supported on the Owens cluster. Intel, PGI and GNU compiler suites are available. The Intel development tool chain is loaded by default. Compiler commands and recommended options for serial programs are listed in the table below. See also our compilation guide.
The Haswell and Broadwell processors that make up Owens support the Advanced Vector Extensions (AVX2) instruction set, but you must set the correct compiler flags to take advantage of it. AVX2 has the potential to speed up your code by a factor of 4 or more, depending on the compiler and options you would otherwise use.
In our experience, the Intel and PGI compilers do a much better job than the GNU compilers at optimizing HPC code.
With the Intel compilers, use -xHost and -O2 or higher. With the GNU compilers, use -march=native and -O3. The PGI compilers by default use the highest available instruction set, so no additional flags are necessary.
This advice assumes that you are building and running your code on Owens. The executables will not be portable. Of course, any highly optimized builds, such as those employing the options above, should be thoroughly validated for correctness.
| LANGUAGE | INTEL | GNU | PGI |
|---|---|---|---|
| C | icc -O2 -xHost hello.c | gcc -O3 -march=native hello.c | pgcc -fast hello.c |
| Fortran 77/90 | ifort -O2 -xHost hello.F | gfortran -O3 -march=native hello.F | pgfortran -fast hello.F |
| C++ | icpc -O2 -xHost hello.cpp | g++ -O3 -march=native hello.cpp | pgc++ -fast hello.cpp |
Parallel Programming
MPI
OSC systems use the MVAPICH2 implementation of the Message Passing Interface (MPI), optimized for the high-speed Infiniband interconnect. MPI is a standard library for performing parallel processing using a distributed-memory model. For more information on building your MPI codes, please visit the MPI Library documentation.
MPI programs are started with the srun command. For example,
#!/bin/bash
#SBATCH --nodes=2
srun [ options ] mpi_prog
The srun command will normally spawn one MPI process per task requested in a Slurm batch job. Use the -n ntasks and/or --ntasks-per-node=n option to change that behavior. For example,
#!/bin/bash #SBATCH --nodes=2 # Use the maximum number of CPUs of two nodes srun ./mpi_prog # Run 8 processes per node srun -n 16 --ntasks-per-node=8 ./mpi_prog
The table below shows some commonly used options. Use srun -help for more information.
| OPTION | COMMENT |
|---|---|
-n, --ntasks=ntasks |
total number of tasks to run |
--ntasks-per-node=n |
number of tasks to invoke on each node |
-help |
Get a list of available options |
srun in any circumstances.OpenMP
The Intel, GNU and PGI compilers understand the OpenMP set of directives, which support multithreaded programming. For more information on building OpenMP codes on OSC systems, please visit the OpenMP documentation.
An OpenMP program by default will use a number of threads equal to the number of CPUs requested in a Slurm batch job. To use a different number of threads, set the environment variable OMP_NUM_THREADS. For example,
#!/bin/bash #SBATCH --ntasks=8 # Run 8 threads ./omp_prog # Run 4 threads export OMP_NUM_THREADS=4 ./omp_prog
To run a OpenMP job on an exclusive node:
#!/bin/bash #SBATCH --nodes=1 #SBATCH --exclusive export OMP_NUM_THREADS=$SLURM_CPUS_ON_NODE ./omp_prog
Interactive job only
See the section on interactive batch in batch job submission for details on submitting an interactive job to the cluster.
Hybrid (MPI + OpenMP)
An example of running a job for hybrid code:
#!/bin/bash #SBATCH --nodes=2 # Run 4 MPI processes on each node and 7 OpenMP threads spawned from a MPI process export OMP_NUM_THREADS=7 srun -n 8 -c 7 --ntasks-per-node=4 ./hybrid_prog
Tuning Parallel Program Performance: Process/Thread Placement
To get the maximum performance, it is important to make sure that processes/threads are located as close as possible to their data, and as close as possible to each other if they need to work on the same piece of data, with given the arrangement of node, sockets, and cores, with different access to RAM and caches.
While cache and memory contention between threads/processes are an issue, it is best to use scatter distribution for code.
Processes and threads are placed differently depending on the computing resources you requste and the compiler and MPI implementation used to compile your code. For the former, see the above examples to learn how to run a job on exclusive nodes. For the latter, this section summarizes the default behavior and how to modify placement.
OpenMP only
For all three compilers (Intel, GNU, PGI), purely threaded codes do not bind to particular CPU cores by default. In other words, it is possible that multiple threads are bound to the same CPU core.
The following table describes how to modify the default placements for pure threaded code:
| DISTRIBUTION | Compact | Scatter/Cyclic |
|---|---|---|
| DESCRIPTION | Place threads as closely as possible on sockets | Distribute threads as evenly as possible across sockets |
| INTEL | KMP_AFFINITY=compact | KMP_AFFINITY=scatter |
| GNU | OMP_PLACES=sockets[1] | OMP_PROC_BIND=spread/close |
| PGI[2] |
MP_BIND=yes |
MP_BIND=yes |
- Threads in the same socket might be bound to the same CPU core.
- PGI LLVM-backend (version 19.1 and later) does not support thread/processors affinity on NUMA architecture. To enable this feature, compile threaded code with
--Mnollvmto use proprietary backend.
MPI Only
For MPI-only codes, MVAPICH2 first binds as many processes as possible on one socket, then allocates the remaining processes on the second socket so that consecutive tasks are near each other. Intel MPI and OpenMPI alternately bind processes on socket 1, socket 2, socket 1, socket 2 etc, as cyclic distribution.
For process distribution across nodes, all MPIs first bind as many processes as possible on one node, then allocates the remaining processes on the second node.
The following table describe how to modify the default placements on a single node for MPI-only code with the command srun:
| DISTRIBUTION (single node) |
Compact | Scatter/Cyclic |
|---|---|---|
| DESCRIPTION | Place processs as closely as possible on sockets | Distribute process as evenly as possible across sockets |
| MVAPICH2[1] | Default | MV2_CPU_BINDING_POLICY=scatter |
| INTEL MPI | srun --cpu-bind="map_cpu:$(seq -s, 0 2 27),$(seq -s, 1 2 27)" | Default |
| OPENMPI | srun --cpu-bind="map_cpu:$(seq -s, 0 2 27),$(seq -s, 1 2 27)" | Default |
MV2_CPU_BINDING_POLICYwill not work ifMV2_ENABLE_AFFINITY=0is set.
To distribute processes evenly across nodes, please set SLURM_DISTRIBUTION=cyclic.
Hybrid (MPI + OpenMP)
For Hybrid codes, each MPI process is allocated OMP_NUM_THREADS cores and the threads of each process are bound to those cores. All MPI processes (as well as the threads bound to the process) behave as we describe in the previous sections. It means the threads spawned from a MPI process might be bound to the same core. To change the default process/thread placmements, please refer to the tables above.
Summary
The above tables list the most commonly used settings for process/thread placement. Some compilers and Intel libraries may have additional options for process and thread placement beyond those mentioned on this page. For more information on a specific compiler/library, check the more detailed documentation for that library.
GPU Programming
160 Nvidia P100 GPUs are available on Owens. Please visit our GPU documentation.
Reference
Citation
For more information about citations of OSC, visit https://www.osc.edu/citation.
To cite Owens, please use the following Archival Resource Key:
ark:/19495/hpc6h5b1
Please adjust this citation to fit the citation style guidelines required.
Ohio Supercomputer Center. 2016. Owens Supercomputer. Columbus, OH: Ohio Supercomputer Center. http://osc.edu/ark:19495/hpc6h5b1
Here is the citation in BibTeX format:
@misc{Owens2016,
ark = {ark:/19495/hpc93fc8},
url = {http://osc.edu/ark:/19495/hpc6h5b1},
year = {2016},
author = {Ohio Supercomputer Center},
title = {Owens Supercomputer}
}
And in EndNote format:
%0 Generic %T Owens Supercomputer %A Ohio Supercomputer Center %R ark:/19495/hpc6h5b1 %U http://osc.edu/ark:/19495/hpc6h5b1 %D 2016
Here is an .ris file to better suit your needs. Please change the import option to .ris.
Owens SSH key fingerprints
These are the public key fingerprints for Owens:
owens: ssh_host_rsa_key.pub = 18:68:d4:b0:44:a8:e2:74:59:cc:c8:e3:3a:fa:a5:3f
owens: ssh_host_ed25519_key.pub = 1c:3d:f9:99:79:06:ac:6e:3a:4b:26:81:69:1a:ce:83
owens: ssh_host_ecdsa_key.pub = d6:92:d1:b0:eb:bc:18:86:0c:df:c5:48:29:71:24:af
These are the SHA256 hashes:
owens: ssh_host_rsa_key.pub = SHA256:vYIOstM2e8xp7WDy5Dua1pt/FxmMJEsHtubqEowOaxo
owens: ssh_host_ed25519_key.pub = SHA256:FSb9ZxUoj5biXhAX85tcJ/+OmTnyFenaSy5ynkRIgV8
owens: ssh_host_ecdsa_key.pub = SHA256:+fqAIqaMW/DUJDB0v/FTxMT9rkbvi/qVdMKVROHmAP4
Batch Limit Rules
Memory Limit:
A small portion of the total physical memory on each node is reserved for distributed processes. The actual physical memory available to user jobs is tabulated below.
Summary
| Node type | default and max memory per core | max memory per node |
|---|---|---|
| regular compute | 4.214 GB | 117 GB |
| huge memory | 31.104 GB | 1492 GB |
| gpu | 4.214 GB | 117 GB |
e.g. The following slurm directives will actually grant this job 3 cores, with 10 GB of memory
(since 2 cores * 4.2 GB = 8.4 GB doesn't satisfy the memory request).
#SBATCH --ntask=2
#SBATCH --mem=10gIt is recommended to let the default memory apply unless more control over memory is needed.
Note that if an entire node is requested, then the job is automatically granted the entire node's main memory. On the other hand, if a partial node is requested, then memory is granted based on the default memory per core.
See a more detailed explanation below.
Regular Dense Compute Node
On Owens, it equates to 4,315 MB/core or 120,820 MB/node (117.98 GB/node) for the regular dense compute node.
If your job requests less than a full node ( ntasks< 28 ), it may be scheduled on a node with other running jobs. In this case, your job is entitled to a memory allocation proportional to the number of cores requested (4315 MB/core). For example, without any memory request ( mem=XXMB ), a job that requests --nodes=1 --ntasks=1 will be assigned one core and should use no more than 4315 MB of RAM, a job that requests --nodes=1 --ntasks=3 will be assigned 3 cores and should use no more than 3*4315 MB of RAM, and a job that requests --nodes=1 --ntasks=28 will be assigned the whole node (28 cores) with 118 GB of RAM.
Here is some information when you include memory request (mem=XX ) in your job. A job that requests --nodes=1 --ntasks=1 --mem=12GB will be assigned three cores and have access to 12 GB of RAM, and charged for 3 cores worth of usage (in other ways, the request --ntasks is ingored). A job that requests --nodes=1 --ntasks=5 --mem=12GB will be assigned 5 cores but have access to only 12 GB of RAM, and charged for 5 cores worth of usage.
A multi-node job ( nodes>1 ) will be assigned the entire nodes with 118 GB/node and charged for the entire nodes regardless of ppn request. For example, a job that requests --nodes=10 --ntasks-per-node=1 will be charged for 10 whole nodes (28 cores/node*10 nodes, which is 280 cores worth of usage).
Huge Memory Node
On Owens, it equates to 31,850 MB/core or 1,528,800 MB/node (1,492.96 GB/node) for a huge memory node.
To request no more than a full huge memory node, you have two options:
- The first is to specify the memory request between 120,832 MB (118 GB) and 1,528,800 MB (1,492.96 GB), i.e.,
120832MB <= mem <=1528800MB(118GB <= mem < 1493GB). Note: you can only use interger for request - The other option is to use the combination of
--ntasks-per-nodeand--partition, like--ntasks-per-node=4 --partition=hugemem. When no memory is specified for the huge memory node, your job is entitled to a memory allocation proportional to the number of cores requested (31,850MB/core). Note,--ntasks-per-nodeshould be no less than 4 and no more than 48.
To manage and monitor your memory usage, please refer to Out-of-Memory (OOM) or Excessive Memory Usage.
GPU Jobs
There is only one GPU per GPU node on Owens.
For serial jobs, we allow node sharing on GPU nodes so a job may request any number of cores (up to 28)
(--nodes=1 --ntasks=XX --gpus-per-node=1)
For parallel jobs (n>1), we do not allow node sharing.
See this GPU computing page for more information.
Partition time and job size limits
Here are the partitions available on Owens:
| Name | Max time limit (dd-hh:mm:ss) |
Min job size | Max job size | notes |
|---|---|---|---|---|
|
serial |
7-00:00:00 |
1 core |
1 node |
|
|
longserial |
14-00:00:0 |
1 core |
1 node |
|
|
parallel |
4-00:00:00 |
2 nodes |
81 nodes |
|
| gpuserial | 7-00:00:00 | 1 core | 1 node | |
| gpuparallel | 4-00:00:00 | 2 nodes | 8 nodes | |
|
hugemem |
7-00:00:00 |
1 core |
1 node |
|
| hugemem-parallel | 4-00:00:00 | 2 nodes | 16 nodes |
|
| debug | 1:00:00 | 1 core | 2 nodes |
|
| gpudebug | 1:00:00 | 1 core | 2 nodes |
|
--partition=<partition-name> to the sbatch command at submission time or add this line to the job script:#SBATCH --paritition=<partition-name>To access one of the restricted queues, please contact OSC Help. Generally, access will only be granted to these queues if the performance of the job cannot be improved, and job size cannot be reduced by splitting or checkpointing the job.
Job/Core Limits
| Max Running Job Limit | Max Core/Processor Limit | ||||
|---|---|---|---|---|---|
| For all types | GPU jobs | Regular debug jobs | GPU debug jobs | For all types | |
| Individual User | 384 | 132 | 4 | 4 | 3080 |
| Project/Group | 576 | 132 | n/a | n/a | 3080 |
An individual user can have up to the max concurrently running jobs and/or up to the max processors/cores in use.
However, among all the users in a particular group/project, they can have up to the max concurrently running jobs and/or up to the max processors/cores in use.
Pitzer
OSC's original Pitzer cluster was installed in late 2018 and is a Dell-built, Intel® Xeon® 'Skylake' processor-based supercomputer with 260 nodes.
In September 2020, OSC installed additional 398 Intel® Xeon® 'Cascade Lake' processor-based nodes as part of a Pitzer Expansion cluster.
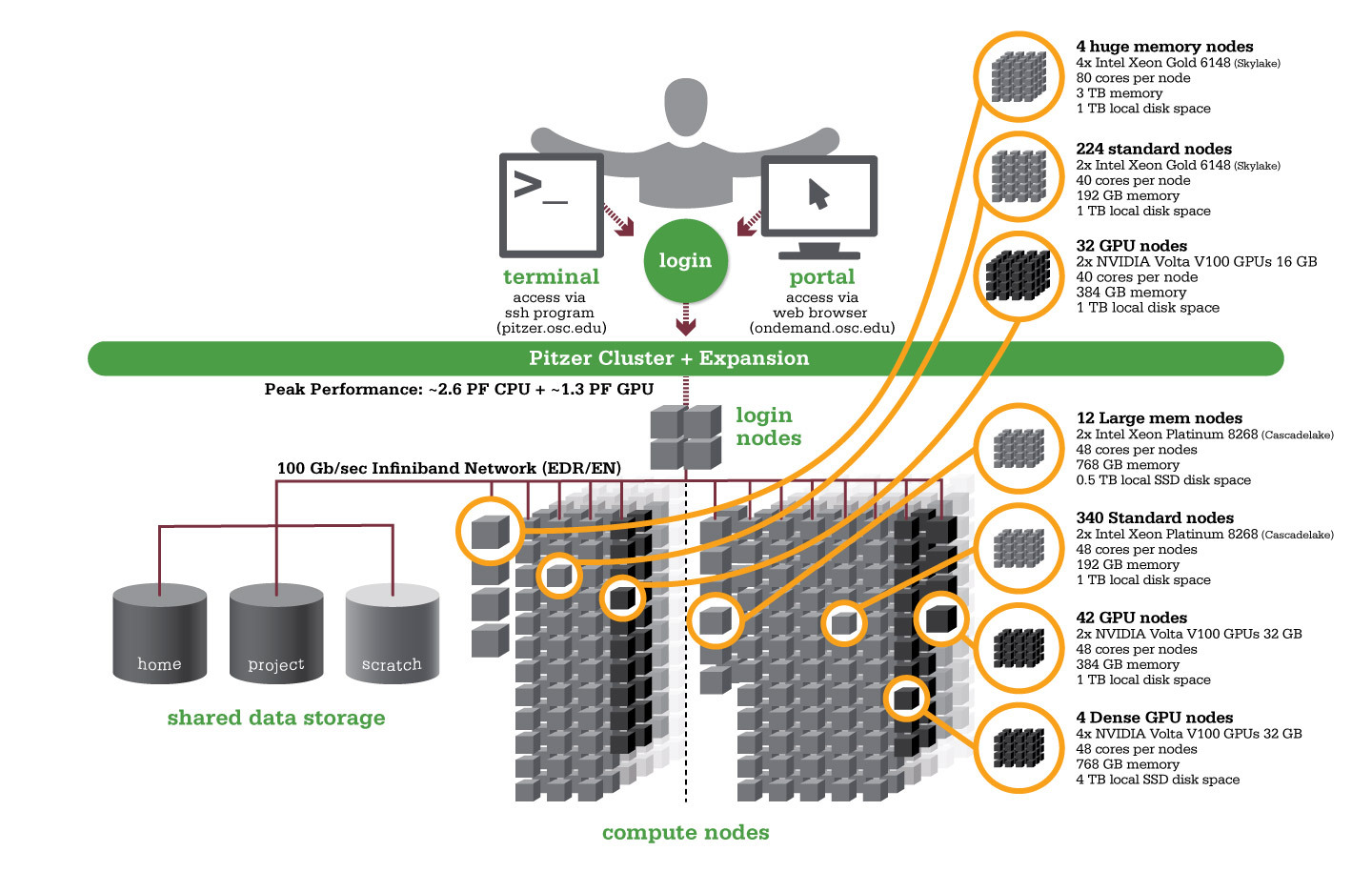
Hardware

Detailed system specifications:
| Deployed in 2018 | Deployed in 2020 | Total | |
|---|---|---|---|
| Total Compute Nodes | 260 Dell nodes | 398 Dell nodes | 658 Dell nodes |
| Total CPU Cores | 10,560 total cores | 19,104 total cores | 29,664 total cores |
| Standard Dense Compute Nodes |
224 nodes
|
340 nodes
|
564 nodes |
| Dual GPU Compute Nodes | 32 nodes
|
42 nodes
|
74 dual GPU nodes |
| Quad GPU Compute Nodes | N/A | 4 nodes
|
4 quad GPU nodes |
| Large Memory Compute Nodes | 4 nodes
|
12 nodes
|
16 nodes |
| Interactive Login Nodes |
4 nodes
|
4 nodes | |
| InfiniBand High-Speed Network | Mellanox EDR (100 Gbps) Infiniband networking | Mellanox EDR (100 Gbps) Infiniband networking | |
| Theoretical Peak Performance |
~850 TFLOPS (CPU only) ~450 TFLOPS (GPU only) ~1300 TFLOPS (total) |
~1900 TFLOPS (CPU only) ~700 TFLOPS (GPU only) ~2600 TFLOPS (total) |
~2750 TFLOPS (CPU only) ~1150 TFLOPS (GPU only) ~3900 TFLOPS (total) |
How to Connect
-
SSH Method
To login to Pitzer at OSC, ssh to the following hostname:
pitzer.osc.edu
You can either use an ssh client application or execute ssh on the command line in a terminal window as follows:
ssh <username>@pitzer.osc.edu
You may see a warning message including SSH key fingerprint. Verify that the fingerprint in the message matches one of the SSH key fingerprints listed here, then type yes.
From there, you are connected to the Pitzer login node and have access to the compilers and other software development tools. You can run programs interactively or through batch requests. We use control groups on login nodes to keep the login nodes stable. Please use batch jobs for any compute-intensive or memory-intensive work. See the following sections for details.
-
OnDemand Method
You can also login to Pitzer at OSC with our OnDemand tool. The first step is to log into OnDemand. Then once logged in you can access Pitzer by clicking on "Clusters", and then selecting ">_Pitzer Shell Access".
Instructions on how to connect to OnDemand can be found at the OnDemand documentation page.
File Systems
Pitzer accesses the same OSC mass storage environment as our other clusters. Therefore, users have the same home directory as on the old clusters. Full details of the storage environment are available in our storage environment guide.
Software Environment
The module system on Pitzer is the same as on the Owens and Ruby systems. Use module load <package> to add a software package to your environment. Use module list to see what modules are currently loaded and module avail to see the modules that are available to load. To search for modules that may not be visible due to dependencies or conflicts, use module spider . By default, you will have the batch scheduling software modules, the Intel compiler, and an appropriate version of mvapich2 loaded.
You can keep up to the software packages that have been made available on Pitzer by viewing the Software by System page and selecting the Pitzer system.
Compiling Code to Use Advanced Vector Extensions (AVX2)
The Skylake processors that make Pitzer support the Advanced Vector Extensions (AVX2) instruction set, but you must set the correct compiler flags to take advantage of it. AVX2 has the potential to speed up your code by a factor of 4 or more, depending on the compiler and options you would otherwise use.
In our experience, the Intel and PGI compilers do a much better job than the gnu compilers at optimizing HPC code.
With the Intel compilers, use -xHost and -O2 or higher. With the gnu compilers, use -march=native and -O3 . The PGI compilers by default use the highest available instruction set, so no additional flags are necessary.
This advice assumes that you are building and running your code on Pitzer. The executables will not be portable. Of course, any highly optimized builds, such as those employing the options above, should be thoroughly validated for correctness.
See the Pitzer Programming Environment page for details.
Batch Specifics
Refer to this Slurm migration page to understand how to use Slurm on the Pitzer cluster. Some specifics you will need to know to create well-formed batch scripts:
- OSC enables PBS compatibility layer provided by Slurm such that PBS batch scripts that used to work in the previous Torque/Moab environment mostly still work in Slurm.
- Pitzer is a heterogeneous system with mixed types of CPUs after the expansion as shown in the above table. Please be cautious when requesting resources on Pitzer and check this page for more detailed discussions
- Jobs on Pitzer may request partial nodes.
Using OSC Resources
For more information about how to use OSC resources, please see our guide on batch processing at OSC and Slurm migration. For specific information about modules and file storage, please see the Batch Execution Environment page.
Technical Specifications
- Login Specifications
-
4 Intel Xeon Gold 6148 (Skylake) CPUs
- 40 cores/node and 384 GB of memory/node
Technical specifications for 2018 Pitzer:
- Number of Nodes
-
260 nodes
- Number of CPU Sockets
-
528 (2 sockets/node for standard node)
- Number of CPU Cores
-
10,560 (40 cores/node for standard node)
- Cores Per Node
-
40 cores/node (80 cores/node for Huge Mem Nodes)
- Local Disk Space Per Node
-
850 GB in /tmp
- Compute CPU Specifications
-
Intel Xeon Gold 6148 (Skylake) for compute
- 2.4 GHz
- 20 cores per processor
- Computer Server Specifications
-
- 224 Dell PowerEdge C6420
- 32 Dell PowerEdge R740 (for accelerator nodes)
- 4 Dell PowerEdge R940
- Accelerator Specifications
-
NVIDIA V100 "Volta" GPUs 16GB memory
- Number of Accelerator Nodes
-
32 total (2 GPUs per node)
- Total Memory
-
~67 TB
- Memory Per Node
-
- 192 GB for standard nodes
- 384 GB for accelerator nodes
- 3 TB for Huge Mem Nodes
- Memory Per Core
-
- 4.8 GB for standard nodes
- 9.6 GB for accelerator nodes
- 76.8 GB for Huge Mem
- Interconnect
-
Mellanox EDR Infiniband Networking (100Gbps)
-
- Special Nodes
-
4 Huge Memory Nodes
- Dell PowerEdge R940
- 4 Intel Xeon Gold 6148 (Skylake)
- 20 Cores
- 2.4 GHz
- 80 cores (20 cores/CPU)
- 3 TB Memory
- 2x Mirror 1 TB Drive (1 TB usable)
Technical specifications for 2020 Pitzer:
- Number of Nodes
-
398 nodes
- Number of CPU Sockets
-
796 (2 sockets/node for all nodes)
- Number of CPU Cores
-
19,104 (48 cores/node for all nodes)
- Cores Per Node
-
48 cores/node for all nodes
- Local Disk Space Per Node
-
- 1 TB for most nodes
- 4 TB for quad GPU
- 0.5 TB for large mem
- Compute CPU Specifications
-
Intel Xeon 8268s Cascade Lakes for most compute
- 2.9 GHz
- 24 cores per processor
- Computer Server Specifications
-
- 352 Dell PowerEdge C6420
- 42 Dell PowerEdge R740 (for dual GPU nodes)
- 4 Dell Poweredge c4140 (for quad GPU nodes)
- Accelerator Specifications
-
- NVIDIA V100 "Volta" GPUs 32GB memory for dual GPU
- NVIDIA V100 "Volta" GPUs 32GB memory and NVLink for quad GPU
- Number of Accelerator Nodes
-
- 42 dual GPU nodes (2 GPUs per node)
- 4 quad GPU nodes (4 GPUs per node)
- Total Memory
-
~95 TB
- Memory Per Node
-
- 192 GB for standard nodes
- 384 GB for dual GPU nodes
- 768 GB for quad and Large Mem Nodes
- Memory Per Core
-
- 4.0 GB for standard nodes
- 8.0 GB for dual GPU nodes
- 16.0 GB for quad and Large Mem Nodes
- Interconnect
-
Mellanox EDR Infiniband Networking (100Gbps)
- Special Nodes
-
4 quad GPU Nodes
- Dual Intel Xeon 8260s Cascade Lakes
- Quad NVIDIA Volta V100s w/32GB GPU memory and NVLink
- 48 cores per node @ 2.4GHz
- 768GB memory
- 4 TB disk space
12 Large Memory Nodes- Dual Intel Xeon 8268 Cascade Lakes
- 48 cores per node @ 2.9GHz
- 768GB memory
- 0.5 TB disk space
Pitzer Programming Environment (PBS)
This document is obsoleted and kept as a reference to previous Pitzer programming environment. Please refer to here for the latest version.
Compilers
C, C++ and Fortran are supported on the Pitzer cluster. Intel, PGI and GNU compiler suites are available. The Intel development tool chain is loaded by default. Compiler commands and recommended options for serial programs are listed in the table below. See also our compilation guide.
The Skylake processors that make up Pitzer support the Advanced Vector Extensions (AVX512) instruction set, but you must set the correct compiler flags to take advantage of it. AVX512 has the potential to speed up your code by a factor of 8 or more, depending on the compiler and options you would otherwise use. However, bare in mind that clock speeds decrease as the level of the instruction set increases. So, if your code does not benefit from vectorization it may be beneficial to use a lower instruction set.
In our experience, the Intel and PGI compilers do a much better job than the GNU compilers at optimizing HPC code.
With the Intel compilers, use -xHost and -O2 or higher. With the GNU compilers, use -march=native and -O3. The PGI compilers by default use the highest available instruction set, so no additional flags are necessary.
This advice assumes that you are building and running your code on Owens. The executables will not be portable. Of course, any highly optimized builds, such as those employing the options above, should be thoroughly validated for correctness.
| LANGUAGE | INTEL EXAMPLE | PGI EXAMPLE | GNU EXAMPLE |
|---|---|---|---|
| C | icc -O2 -xHost hello.c | pgcc -fast hello.c | gcc -O3 -march=native hello.c |
| Fortran 90 | ifort -O2 -xHost hello.f90 | pgf90 -fast hello.f90 | gfortran -O3 -march=native hello.f90 |
| C++ | icpc -O2 -xHost hello.cpp | pgc++ -fast hello.cpp | g++ -O3 -march=native hello.cpp |
Parallel Programming
MPI
OSC systems use the MVAPICH2 implementation of the Message Passing Interface (MPI), optimized for the high-speed Infiniband interconnect. MPI is a standard library for performing parallel processing using a distributed-memory model. For more information on building your MPI codes, please visit the MPI Library documentation.
Parallel programs are started with the mpiexec command. For example,
mpiexec ./myprog
The mpiexec command will normally spawn one MPI process per CPU core requested in a batch job. Use the -n and/or -ppn option to change that behavior.
The table below shows some commonly used options. Use mpiexec -help for more information.
| MPIEXEC OPTION | COMMENT |
|---|---|
-ppn 1 |
One process per node |
-ppn procs |
procs processes per node |
-n totalprocs-np totalprocs |
At most totalprocs processes per node |
-prepend-rank |
Prepend rank to output |
-help |
Get a list of available options |
OpenMP
The Intel, PGI and GNU compilers understand the OpenMP set of directives, which support multithreaded programming. For more information on building OpenMP codes on OSC systems, please visit the OpenMP documentation.
Process/Thread placement
Processes and threads are placed differently depending on the compiler and MPI implementation used to compile your code. This section summarizes the default behavior and how to modify placement.
For all three compilers (Intel, GNU, PGI), purely threaded codes do not bind to particular cores by default.
For MPI-only codes, Intel MPI first binds the first half of processes to one socket, and then second half to the second socket so that consecutive tasks are located near each other. MVAPICH2 first binds as many processes as possible on one socket, then allocates the remaining processes on the second socket so that consecutive tasks are near each other. OpenMPI alternately binds processes on socket 1, socket 2, socket 1, socket 2, etc, with no particular order for the core id.
For Hybrid codes, Intel MPI first binds the first half of processes to one socket, and then second half to the second socket so that consecutive tasks are located near each other. Each process is allocated ${OMP_NUM_THREADS} cores and the threads of each process are bound to those cores. MVAPICH2 allocates ${OMP_NUM_THREADS} cores for each process and each thread of a process is placed on a separate core. By default, OpenMPI behaves the same for hybrid codes as it does for MPI-only codes, allocating a single core for each process and all threads of that process.
The following tables describe how to modify the default placements for each type of code.
OpenMP options:
| Option | Intel | GNU | Pgi | description |
|---|---|---|---|---|
| Scatter | KMP_AFFINITY=scatter | OMP_PLACES=cores OMP_PROC_BIND=close/spread | MP_BIND=yes | Distribute threads as evenly as possible across system |
| Compact | KMP_AFFINITY=compact | OMP_PLACES=sockets | MP_BIND=yes MP_BLIST="0,2,4,6,8,10,1,3,5,7,9" | Place threads as closely as possible on system |
MPI options:
| OPTION | INTEL | MVAPICh2 | openmpi | DESCRIPTION |
|---|---|---|---|---|
| Scatter | I_MPI_PIN_DOMAIN=core I_MPI_PIN_ORDER=scatter | MV2_CPU_BINDING_POLICY=scatter | -map-by core --rank-by socket:span | Distribute processes as evenly as possible across system |
| Compact | I_MPI_PIN_DOMAIN=core I_MPI_PIN_ORDER=compact | MV2_CPU_BINDING_POLICY=bunch | -map-by core |
Distribute processes as closely as possible on system |
Hybrid MPI+OpenMP options (combine with options from OpenMP table for thread affinity within cores allocated to each process):
| OPTION | INTEL | MVAPICH2 | OPENMPI | DESCRIPTION |
|---|---|---|---|---|
| Scatter | I_MPI_PIN_DOMAIN=omp I_MPI_PIN_ORDER=scatter | MV2_CPU_BINDING_POLICY=hybrid MV2_HYBRID_BINDING_POLICY=linear | -map-by node:PE=$OMP_NUM_THREADS --bind-to core --rank-by socket:span | Distrubute processes as evenly as possible across system ($OMP_NUM_THREADS cores per process) |
| Compact | I_MPI_PIN_DOMAIN=omp I_MPI_PIN_ORDER=compact | MV2_CPU_BINDING_POLICY=hybrid MV2_HYBRID_BINDING_POLICY=spread | -map-by node:PE=$OMP_NUM_THREADS --bind-to core | Distribute processes as closely as possible on system ($OMP_NUM_THREADS cores per process) |
The above tables list the most commonly used settings for process/thread placement. Some compilers and Intel libraries may have additional options for process and thread placement beyond those mentioned on this page. For more information on a specific compiler/library, check the more detailed documentation for that library.
GPU Programming
64 Nvidia V100 GPUs are available on Pitzer. Please visit our GPU documentation.
Pitzer Programming Environment
Compilers
C, C++ and Fortran are supported on the Pitzer cluster. Intel, PGI and GNU compiler suites are available. The Intel development tool chain is loaded by default. Compiler commands and recommended options for serial programs are listed in the table below. See also our compilation guide.
The Skylake and Cascade Lake processors that make up Pitzer support the Advanced Vector Extensions (AVX512) instruction set, but you must set the correct compiler flags to take advantage of it. AVX512 has the potential to speed up your code by a factor of 8 or more, depending on the compiler and options you would otherwise use. However, bare in mind that clock speeds decrease as the level of the instruction set increases. So, if your code does not benefit from vectorization it may be beneficial to use a lower instruction set.
In our experience, the Intel compiler usually does the best job of optimizing numerical codes and we recommend that you give it a try if you’ve been using another compiler.
With the Intel compilers, use -xHost and -O2 or higher. With the GNU compilers, use -march=native and -O3. The PGI compilers by default use the highest available instruction set, so no additional flags are necessary.
This advice assumes that you are building and running your code on Owens. The executables will not be portable. Of course, any highly optimized builds, such as those employing the options above, should be thoroughly validated for correctness.
| LANGUAGE | INTEL | GNU | PGI |
|---|---|---|---|
| C | icc -O2 -xHost hello.c | gcc -O3 -march=native hello.c | pgcc -fast hello.c |
| Fortran 77/90 | ifort -O2 -xHost hello.F | gfortran -O3 -march=native hello.F | pgfortran -fast hello.F |
| C++ | icpc -O2 -xHost hello.cpp | g++ -O3 -march=native hello.cpp | pgc++ -fast hello.cpp |
Parallel Programming
MPI
OSC systems use the MVAPICH2 implementation of the Message Passing Interface (MPI), optimized for the high-speed Infiniband interconnect. MPI is a standard library for performing parallel processing using a distributed-memory model. For more information on building your MPI codes, please visit the MPI Library documentation.
MPI programs are started with the srun command. For example,
#!/bin/bash
#SBATCH --nodes=2
srun [ options ] mpi_prog
The srun command will normally spawn one MPI process per task requested in a Slurm batch job. Use the -n ntasks and/or --ntasks-per-node=n option to change that behavior. For example,
#!/bin/bash #SBATCH --nodes=2 # Use the maximum number of CPUs of two nodes srun ./mpi_prog # Run 8 processes per node srun -n 16 --ntasks-per-node=8 ./mpi_prog
The table below shows some commonly used options. Use srun -help for more information.
| OPTION | COMMENT |
|---|---|
-n, --ntasks=ntasks |
total number of tasks to run |
--ntasks-per-node=n |
number of tasks to invoke on each node |
-help |
Get a list of available options |
srun in any circumstances.OpenMP
The Intel, GNU and PGI compilers understand the OpenMP set of directives, which support multithreaded programming. For more information on building OpenMP codes on OSC systems, please visit the OpenMP documentation.
An OpenMP program by default will use a number of threads equal to the number of CPUs requested in a Slurm batch job. To use a different number of threads, set the environment variable OMP_NUM_THREADS. For example,
#!/bin/bash #SBATCH --ntasks=8 # Run 8 threads ./omp_prog # Run 4 threads export OMP_NUM_THREADS=4 ./omp_prog
To run a OpenMP job on an exclusive node:
#!/bin/bash #SBATCH --nodes=1 #SBATCH --exclusive export OMP_NUM_THREADS=$SLURM_CPUS_ON_NODE ./omp_prog
Interactive job only
Please use -c, --cpus-per-task=X instead of -n, --ntasks=X to request an interactive job. Both result in an interactive job with X CPUs available but only the former option automatically assigns the correct number of threads to the OpenMP program. If the option --ntasks is used only, the OpenMP program will use one thread or all threads will be bound to one CPU core.
Hybrid (MPI + OpenMP)
An example of running a job for hybrid code:
#!/bin/bash #SBATCH --nodes=2 #SBATCH --constraint=48core # Run 4 MPI processes on each node and 12 OpenMP threads spawned from a MPI process export OMP_NUM_THREADS=12 srun -n 8 -c 12 --ntasks-per-node=4 ./hybrid_prog
To run a job across either 40-core or 48-core nodes exclusively:
#!/bin/bash #SBATCH --nodes=2 # Run 4 MPI processes on each node and the maximum available OpenMP threads spawned from a MPI process export OMP_NUM_THREADS=$(($SLURM_CPUS_ON_NODE/4)) srun -n 8 -c $OMP_NUM_THREADS --ntasks-per-node=4 ./hybrid_prog
Tuning Parallel Program Performance: Process/Thread Placement
To get the maximum performance, it is important to make sure that processes/threads are located as close as possible to their data, and as close as possible to each other if they need to work on the same piece of data, with given the arrangement of node, sockets, and cores, with different access to RAM and caches.
While cache and memory contention between threads/processes are an issue, it is best to use scatter distribution for code.
Processes and threads are placed differently depending on the computing resources you requste and the compiler and MPI implementation used to compile your code. For the former, see the above examples to learn how to run a job on exclusive nodes. For the latter, this section summarizes the default behavior and how to modify placement.
OpenMP only
For all three compilers (Intel, GNU, PGI), purely threaded codes do not bind to particular CPU cores by default. In other words, it is possible that multiple threads are bound to the same CPU core.
The following table describes how to modify the default placements for pure threaded code:
| DISTRIBUTION | Compact | Scatter/Cyclic |
|---|---|---|
| DESCRIPTION | Place threads as closely as possible on sockets | Distribute threads as evenly as possible across sockets |
| INTEL | KMP_AFFINITY=compact | KMP_AFFINITY=scatter |
| GNU | OMP_PLACES=sockets[1] | OMP_PROC_BIND=spread/close |
| PGI[2] |
MP_BIND=yes |
MP_BIND=yes |
- Threads in the same socket might be bound to the same CPU core.
- PGI LLVM-backend (version 19.1 and later) does not support thread/processors affinity on NUMA architecture. To enable this feature, compile threaded code with
--Mnollvmto use proprietary backend.
MPI Only
For MPI-only codes, MVAPICH2 first binds as many processes as possible on one socket, then allocates the remaining processes on the second socket so that consecutive tasks are near each other. Intel MPI and OpenMPI alternately bind processes on socket 1, socket 2, socket 1, socket 2 etc, as cyclic distribution.
For process distribution across nodes, all MPIs first bind as many processes as possible on one node, then allocates the remaining processes on the second node.
The following table describe how to modify the default placements on a single node for MPI-only code with the command srun:
| DISTRIBUTION (single node) |
Compact | Scatter/Cyclic |
|---|---|---|
| DESCRIPTION | Place processs as closely as possible on sockets | Distribute process as evenly as possible across sockets |
| MVAPICH2[1] | Default | MV2_CPU_BINDING_POLICY=scatter |
| INTEL MPI | srun --cpu-bind="map_cpu:$(seq -s, 0 2 47),$(seq -s, 1 2 47)" | Default |
| OPENMPI | srun --cpu-bind="map_cpu:$(seq -s, 0 2 47),$(seq -s, 1 2 47)" | Default |
MV2_CPU_BINDING_POLICYwill not work ifMV2_ENABLE_AFFINITY=0is set.
To distribute processes evenly across nodes, please set SLURM_DISTRIBUTION=cyclic.
Hybrid (MPI + OpenMP)
For Hybrid codes, each MPI process is allocated OMP_NUM_THREADS cores and the threads of each process are bound to those cores. All MPI processes (as well as the threads bound to the process) behave as we describe in the previous sections. It means the threads spawned from a MPI process might be bound to the same core. To change the default process/thread placmements, please refer to the tables above.
Summary
The above tables list the most commonly used settings for process/thread placement. Some compilers and Intel libraries may have additional options for process and thread placement beyond those mentioned on this page. For more information on a specific compiler/library, check the more detailed documentation for that library.
GPU Programming
164 Nvidia V100 GPUs are available on Pitzer. Please visit our GPU documentation.
Reference
Batch Limit Rules
Memory limit
A small portion of the total physical memory on each node is reserved for distributed processes. The actual physical memory available to user jobs is tabulated below.
Summary
| Node type | default and max memory per core | max memory per node |
|---|---|---|
| Skylake 40 core - regular compute | 4.449 GB | 177.96 GB |
| Cascade Lake 48 core - regular compute | 3.708 GB | 177.98 GB |
| large memory | 15.5 GB | 744 GB |
| huge memory | 37.362 GB | 2988.98 GB |
| Skylake 40 core dual gpu | 9.074 GB | 363 GB |
| Cascade 48 core dual gpu | 7.562 GB | 363 GB |
| quad gpu (48 core) | 15.5 GB |
744 GB |
A job may request more than the max memory per core, but the job will be allocated more cores to satisfy the memory request instead of just more memory.
e.g. The following slurm directives will actually grant this job 3 cores, with 10 GB of memory
(since 2 cores * 4.5 GB = 9 GB doesn't satisfy the memory request).#SBATCH --ntask=2
#SBATCH --mem=10g
It is recommended to let the default memory apply unless more control over memory is needed.
Note that if an entire node is requested, then the job is automatically granted the entire node's main memory. On the other hand, if a partial node is requested, then memory is granted based on the default memory per core.
See a more detailed explanation below.
Regular Compute Node
- For the regular 'Skylake' processor-based node, it has 40 cores/node. The physical memory equates to 4.8 GB/core or 192 GB/node; while the usable memory equates to 4,556 MB/core or 182,240 MB/node (177.96 GB/node).
- For the regular 'Cascade Lake' processor-based node, it has 48 cores/node. The physical memory equates to 4.0 GB/core or 192 GB/node; while the usable memory equates to 3,797 MB/core or 182,256 MB/node (177.98 GB/node).
Jobs requesting no more than 1 node
If your job requests less than a full node, it may be scheduled on a node with other running jobs. In this case, your job is entitled to a memory allocation proportional to the number of cores requested (4,556 MB/core or 3,797 MB/core depending on which type of node your job lands on). For example, without any memory request ( --mem=XX ):
- A job that requests
--ntasks=1and lands on a 'Skylake' node will be assigned one core and should use no more than 4556 MB of RAM; a job that requests--ntasks=1and lands on a 'Cascade Lake' node will be assigned one core and should use no more than 3797 MB of RAM - A job that requests
--ntasks=3and lands on a 'Skylake' node will be assigned 3 cores and should use no more than 3*4556 MB of RAM; a job that requests--ntasks=3and lands on a 'Cascade Lake' node will be assigned 3 cores and should use no more than 3*3797 MB of RAM - A job that requests
--ntasks=40and lands on a 'Skylake' node will be assigned the whole node (40 cores) with 178 GB of RAM; a job that requests--ntasks=40and lands on a 'Cascade Lake' node will be assigned 40 cores (partial node) and should use no more than 40* 3797 MB of RAM - A job that requests
--exclusiveand lands on a 'Skylake' node will be assigned the whole node (40 cores) with 178 GB of RAM; a job that requests--exclusiveand lands on a 'Cascade Lake' node will be assigned the whole node (48 cores) with 178 GB of RAM - A job that requests
--exclusive --constraint=40corewill land on a 'Skylake' node and will be assigned the whole node (40 cores) with 178 GB of RAM.
For example, with a memory request: - A job that requests
--ntasks=1 --mem=16000MBand lands on 'Skylake' node will be assigned 4 cores and have access to 16000 MB of RAM, and charged for 4 cores worth of usage; a job that requests--ntasks=1 --mem=16000MBand lands on 'Cascade Lake' node will be assigned 5 cores and have access to 16000 MB of RAM, and charged for 5 cores worth of usage - A job that requests
--ntasks=8 --mem=16000MBand lands on 'Skylake' node will be assigned 8 cores but have access to only 16000 MB of RAM , and charged for 8 cores worth of usage; a job that requests--ntasks=8 --mem=16000MBand lands on 'Cascade Lake' node will be assigned 8 cores but have access to only 16000 MB of RAM , and charged for 8 cores worth of usage
Jobs requesting more than 1 node
A multi-node job ( --nodes > 1 ) will be assigned the entire nodes and charged for the entire nodes regardless of --ntasks or --ntasks-per-node request. For example, a job that requests --nodes=10 --ntasks-per-node=1 and lands on 'Skylake' node will be charged for 10 whole nodes (40 cores/node*10 nodes, which is 400 cores worth of usage); a job that requests --nodes=10 --ntasks-per-node=1 and lands on 'Cascade Lake' node will be charged for 10 whole nodes (48 cores/node*10 nodes, which is 480 cores worth of usage). We usually suggest not including --ntasks-per-node and using --ntasks if needed.
Large Memory Node
On Pitzer, it has 48 cores per node. The physical memory equates to 16.0 GB/core or 768 GB/node; while the usable memory equates to 15,872 MB/core or 761,856 MB/node (744 GB/node).
For any job that requests no less than 363 GB/node but less than 744 GB/node, the job will be scheduled on the large memory node.To request no more than a full large memory node, you need to specify the memory request between 363 GB and 744 GB, i.e., 363GB <= mem <744GB. --mem is the total memory per node allocated to the job. You can request a partial large memory node, so consider your request more carefully when you plan to use a large memory node, and specify the memory based on what you will use.
Huge Memory Node
On Pitzer, it has 80 cores per node. The physical memory equates to 37.5 GB/core or 3 TB/node; while the usable memory equates to 38,259 MB/core or 3,060,720 MB/node (2988.98 GB/node).
To request no more than a full huge memory node, you have two options:
- The first is to specify the memory request between 744 GB and 2988 GB, i.e.,
744GB <= mem <=2988GB). - The other option is to use the combination of
--ntasks-per-nodeand--partition, like--ntasks-per-node=4 --partition=hugemem. When no memory is specified for the huge memory node, your job is entitled to a memory allocation proportional to the number of cores requested (38,259 MB/core). Note,--ntasks-per-nodeshould be no less than 20 and no more than 80
Summary
In summary, for serial jobs, we will allocate the resources considering both the # of cores and the memory request. For parallel jobs (nodes>1), we will allocate the entire nodes with the whole memory regardless of other requests. Check requesting resources on pitzer for information about the usable memory of different types of nodes on Pitzer. To manage and monitor your memory usage, please refer to Out-of-Memory (OOM) or Excessive Memory Usage.
GPU Jobs
Dual GPU Node
- For the dual GPU node with 'Skylake' processor, it has 40 cores/node. The physical memory equates to 9.6 GB/core or 384 GB/node; while the usable memory equates to 9292 MB/core or 363 GB/node. Each node has 2 NVIDIA Volta V100 w/ 16 GB GPU memory.
- For the dual GPU node with 'Cascade Lake' processor, it has 48 cores/node. The physical memory equates to 8.0 GB/core or 384 GB/node; while the usable memory equates to 7744 MB/core or 363 GB/node. Each node has 2 NVIDIA Volta V100 w/32GB GPU memory.
For serial jobs, we will allow node sharing on GPU nodes so a job may request either 1 or 2 GPUs (--ntasks=XX --gpus-per-node=1 or --ntasks=XX --gpus-per-node=2)
For parallel jobs (nodes>1), we will not allow node sharing. A job may request 1 or 2 GPUs ( gpus-per-node=1 or gpus-per-node=2 ) but both GPUs will be allocated to the job.
Quad GPU Node
For quad GPU node, it has 48 cores/node. The physical memory equates to 16.0 GB/core or 768 GB/node; while the usable memory equates to 15,872 MB/core or 744 GB/node.. Each node has 4 NVIDIA Volta V100s w/32 GB GPU memory and NVLink.
For serial jobs, we will allow node sharing on GPU nodes, so a job can land on a quad GPU node if it requests 3-4 GPUs per node (--ntasks=XX --gpus-per-node=3 or --ntasks=XX --gpus-per-node=4), or requests quad GPU node explicitly with using --gpus-per-node=v100-quad:4, or gets backfilled with requesting 1-2 GPUs per node with less than 4 hours long.
For parallel jobs (nodes>1), only up to 2 quad GPU nodes can be requested in a single job. We will not allow node sharing and all GPUs will be allocated to the job.
Partition time and job size limits
Here is the walltime and node limits per job for different queues/partitions available on Pitzer:
|
NAME |
MAX TIME LIMIT |
MIN JOB SIZE |
MAX JOB SIZE |
NOTES |
|---|---|---|---|---|
|
serial |
7-00:00:00 |
1 core |
1 node |
|
| longserial | 14-00:00:00 |
1 core |
1 node |
|
|
parallel |
96:00:00 |
2 nodes |
40 nodes |
|
|
hugemem |
7-00:00:00 |
1 core |
1 node |
|
|
largemem |
7-00:00:00 |
1 core |
1 node |
|
|
gpuserial |
7-00:00:00 |
1 core |
1 node |
|
|
gpuparallel |
96:00:00 |
2 nodes |
10 nodes |
|
|
debug |
1:00:00 |
1 core |
2 nodes |
|
|
gpudebug |
1:00:00 |
1 core |
2 nodes |
|
To specify a partition for a job, either add the flag --partition=<partition-name> to the sbatch command at submission time or add this line to the job script:#SBATCH --paritition=<partition-name>
To access one of the restricted queues, please contact OSC Help. Generally, access will only be granted to these queues if the performance of the job cannot be improved, and job size cannot be reduced by splitting or checkpointing the job.
Job/Core Limits
| Max Running Job Limit | Max Core/Processor Limit | ||||
|---|---|---|---|---|---|
| For all types | GPU jobs | Regular debug jobs | GPU debug jobs | For all types | |
| Individual User | 384 | 140 | 4 | 4 | 3240 |
| Project/Group | 576 | 140 | n/a | n/a | 3240 |
An individual user can have up to the max concurrently running jobs and/or up to the max processors/cores in use. However, among all the users in a particular group/project, they can have up to the max concurrently running jobs and/or up to the max processors/cores in use.
Citation
For more information about citations of OSC, visit https://www.osc.edu/citation.
To cite Pitzer, please use the following Archival Resource Key:
ark:/19495/hpc56htp
Please adjust this citation to fit the citation style guidelines required.
Ohio Supercomputer Center. 2018. Pitzer Supercomputer. Columbus, OH: Ohio Supercomputer Center. http://osc.edu/ark:19495/hpc56htp
Here is the citation in BibTeX format:
@misc{Pitzer2018,
ark = {ark:/19495/hpc56htp},
url = {http://osc.edu/ark:/19495/hpc56htp},
year = {2018},
author = {Ohio Supercomputer Center},
title = {Pitzer Supercomputer}
}
And in EndNote format:
%0 Generic %T Pitzer Supercomputer %A Ohio Supercomputer Center %R ark:/19495/hpc56htp %U http://osc.edu/ark:/19495/hpc56htp %D 2018
Here is an .ris file to better suit your needs. Please change the import option to .ris.
Pitzer SSH key fingerprints
These are the public key fingerprints for Pitzer:
pitzer: ssh_host_rsa_key.pub = 8c:8a:1f:67:a0:e8:77:d5:4e:3b:79:5e:e8:43:49:0e
pitzer: ssh_host_ed25519_key.pub = 6d:19:73:8e:b4:61:09:a9:e6:0f:e5:0d:e5:cb:59:0b
pitzer: ssh_host_ecdsa_key.pub = 6f:c7:d0:f9:08:78:97:b8:23:2e:0d:e2:63:e7:ac:93
These are the SHA256 hashes:
pitzer: ssh_host_rsa_key.pub = SHA256:oWBf+YmIzwIp+DsyuvB4loGrpi2ecow9fnZKNZgEVHc
pitzer: ssh_host_ed25519_key.pub = SHA256:zUgn1K3+FK+25JtG6oFI9hVZjVxty1xEqw/K7DEwZdc
pitzer: ssh_host_ecdsa_key.pub = SHA256:8XAn/GbQ0nbGONUmlNQJenMuY5r3x7ynjnzLt+k+W1M
Migrating jobs from other clusters
This page includes a summary of differences to keep in mind when migrating jobs from other clusters to Pitzer.
Guidance for Oakley Users
Guidance for Owens Users
Hardware Specifications
| pitzer (PER NODE) | owens (PER NODE) | ||
|---|---|---|---|
| Regular compute node |
40 cores and 192GB of RAM 48 cores and 192GB of RAM |
28 cores and 125GB of RAM | |
| Huge memory node |
48 cores and 768GB of RAM (12 nodes in this class) 80 cores and 3.0 TB of RAM (4 nodes in this class) |
48 cores and 1.5TB of RAM (16 nodes in this class) |
|
File Systems
Pitzer accesses the same OSC mass storage environment as our other clusters. Therefore, users have the same home directory, project space, and scratch space as on the Owens cluster.
Software Environment
Pitzer uses the same module system as Owens.
Use module load <package> to add a software package to your environment. Use module list to see what modules are currently loaded and module avail to see the modules that are available to load. To search for modules that may not be visible due to dependencies or conflicts, use module spider .
You can keep up to on the software packages that have been made available on Pitzer by viewing the Software by System page and selecting the Pitzer system.
Programming Environment
Like Owens, Pitzer supports three compilers: Intel, PGI, and gnu. The default is Intel. To switch to a different compiler, use module swap intel gnu or module swap intel pgi .
Pitzer also use the MVAPICH2 implementation of the Message Passing Interface (MPI), optimized for the high-speed Infiniband interconnect and support the Advanced Vector Extensions (AVX2) instruction set.
See the Pitzer Programming Environment page for details.
Accounting
Below is a comparison of job limits between Pitzer and Owens:
| PItzer | Owens | |
|---|---|---|
| Per User | Up to 256 concurrently running jobs and/or up to 3240 processors/cores in use | Up to 256 concurrently running jobs and/or up to 3080 processors/cores in use |
| Per group | Up to 384 concurrently running jobs and/or up to 3240 processors/cores in use | Up to 384 concurrently running jobs and/or up to 4620 processors/cores in use |
Please see Queues and Reservations for Pitzer and Batch Limit Rules for more details.
Guidance for Ruby Users
Guidance on Requesting Resources on Pitzer
In late 2018, OSC installed 260 Intel® Xeon® 'Skylake' processor-based nodes as the original Pitzer cluster. In September 2020, OSC installed additional 398 Intel® Xeon® 'Cascade Lake' processor-based nodes as part of a Pitzer Expansion cluster. This expansion makes Pitzer a heterogeneous cluster, which means that the jobs may land on different types of CPU and behaves differently if the user submits the same job script repeatedly to Pitzer but does not request the resources properly. This document provides you some general guidance on how to request resources on Pitzer due to this heterogeneous nature.
Step 1: Identify your job type
| Nodes the job may be allocated on | # of cores per node | Usable Memory | GPU | |
|---|---|---|---|---|
| Jobs requesting standard compute node(s) | Dual Intel Xeon 6148s Skylake @2.4GHz | 40 |
178 GB memory/node 4556 MB memory/core |
N/A |
| Dual Intel Xeon 8268s Cascade Lakes @2.9GHz | 48 |
178 GB memory/node 3797 MB memory/core |
N/A | |
| Jobs requesting dual GPU node(s) |
Dual Intel Xeon 6148s Skylake @2.4GHz |
40 |
363 GB memory/node 9292 MB memory/core |
2 NVIDIA Volta V100 w/ 16GB GPU memory |
| Dual Intel Xeon 8268s Cascade Lakes @2.9GHz | 48 |
363 GB memory/node 7744 MB memory/core |
2 NVIDIA Volta V100 w/32GB GPU memory | |
| Jobs requesting quad GPU node(s) | Dual Intel Xeon 8260s Cascade Lakes @2.4GHz | 48 |
744 GB memory/node 15872 MB memory/core |
4 NVIDIA Volta V100s w/32GB GPU memory and NVLink |
| Jobs requesting large memory node(s) | Dual Intel Xeon 8268s Cascade Lakes @2.9GHz | 48 |
744 GB memory/node 15872 MB memory/core |
N/A |
| Jobs requesting huge memory node(s) | Quad Processor Intel Xeon 6148 Skylakes @2.4GHz | 80 |
2989 GB memory/node 38259 MB memory/core |
N/A |
According to this table,
- If your job requests standard compute node(s) or dual GPU node(s), it can potentially land on different types of nodes and may result in different job performance. Please follow the steps below to determine whether you would like to restrain your job to a certain type of node(s).
- If your job requests quad GPU node(s), large memory node(s), or huge memory node(s), please check pitzer batch limit rules on how to request these special types of resources properly.
Step 2: Perform test
This step is to submit your jobs requesting the same resources to different types of nodes on Pitzer. For your job script is prepared with either PBS syntax or Slurm syntax:
Request 40 or 48 core nodes
#SBATCH --constraint=40core #SBATCH --constraint=48core
Request 16gb, 32gb gpu
#SBATCH --constraint=v100 #SBATCH --constraint=v100-32g --partition=gpuserial-48core
Once the script is ready, submit your jobs to Pitzer and wait till the jobs are completed.
Step 3: Compare the results
Once the jobs are completed, you can compare the job performances in terms of core-hours, gpu-hours, walltime, etc. to determine how your job is sensitive to the type of the nodes. If you would like to restrain your job to land on a certain type of nodes based on the testing, you can add #SBATCH --constraint=. The disadvantage of this is that you may have a longer queue wait time on the system. If you would like to have your jobs scheduled as fast as possible and do not care which type of nodes your job will land on, do not include the constraint in the job request.
GPU Computing
OSC offers GPU computing on all its systems. While GPUs can provide a significant boost in performance for some applications, the computing model is very different from the CPU. This page will discuss some of the ways you can use GPU computing at OSC.
Accessing GPU Resources
To request nodes with a GPU add the --gpus-per-node=x attribute to the directive in your batch script, for example, on Owens:
#SBATCH --gpus-per-node=1
In most cases you'll need to load the cuda module (module load cuda) to make the necessary Nvidia libraries available.
Setting the GPU compute mode (optional)
The GPUs on Owens and Pitzer can be set to different compute modes as listed here. They can be set by adding the following to the GPU specification when using the srun command. By default it is set to shared.
srun --gpu_cmode=exclusive
or
srun --gpu_cmode=shared
The compute mode shared is the default on GPU nodes if a compute mode is not specified. With this compute mode, mulitple CUDA processes on the same GPU device are allowed.
Using GPU-enabled Applications
We have several supported applications that can use GPUs. This includes
- Machine learning / Neural networks
- Molecular mechanics / dynamics
- General mathematics
- Engineering and Quantum Chemistry applications are expected to follow.
Please see the software pages for each application. They have different levels of support for multi-node jobs, cpu/gpu work sharing, and environment set-up.
Libraries with GPU Support
There are a few libraries that provide GPU implementations of commonly used routines. While they mostly hide the details of using a GPU there are still some GPU specifics you'll need to be aware of, e.g. device initialization, threading, and memory allocation. These are available at OSC:
MAGMA
MAGMA is an implementation of BLAS and LAPACK with multi-core (SMP) and GPU support. There are some differences in the API of standard BLAS and LAPACK.
cuBLAS and cuSPARSE
cuBLAS is a highly optimized BLAS from NVIDIA. There are a few versions of this library, from very GPU-specific to nearly transparent. cuSPARSE is a BLAS-like library for sparse matrices.
The MAGMA library is built on cuBLAS.
cuFFT
cuFFT is NVIDIA's Fourier transform library with an API similar to FFTW.
cuDNN
cuDNN is NVIDIA's Deep Neural Network machine learning library. Many ML applications are built on cuDNN.
Direct GPU Programming
GPUs present a different programming model from CPUs so there is a significant time investment in going this route.
OpenACC
OpenACC is a directives-based model similar to OpenMP. Currently this is only supported by the Portland Group C/C++ and Fortran compilers.
OpenCL
OpenCL is a set of libraries and C/C++ compiler extensions supporting GPUs (NVIDIA and AMD) and other hardware accelerators. The CUDA module provides an OpenCL library.
CUDA
CUDA is the standard NVIDIA development environment. In this model explicit GPU code is written in the CUDA C/C++ dialect, compiled with the CUDA compiler NVCC, and linked with a native driver program.About GPU Hardware
Our GPUs span several generations with different capabilites and ease-of-use. Many of the differences won't be visible when using applications or libraries, but some features and applications may not be supported on the older models.
Owens P100
The P100 "Pascal" is a NVIDIA GPU with a compute capability of 6.0. The 6.0 capability includes unified shared CPU/GPU memory -- the GPU now has its own virtual memory capability and can map CPU memory into its address space.
Each P100 has 16GB of on-board memory and there is one GPU per GPU node.
Pitzer V100
The NVIDIA V100 "Volta" GPU, with a compute capability of 7.0, offers several advanced features, one of which is its Tensor Cores. These Tensor Cores empower the GPU to perform mixed-precision matrix operations, significantly enhancing its efficiency for deep learning workloads and expediting tasks such as AI model training and inference.
The V100 deployed in 2018 comes equipped with 16GB of memory, whereas the V100 deployed in 2020 features 32GB of memory. There are two GPUs per GPU node,
Additionally, there are four large memory nodes equipped with quad NVIDIA Volta V100s with 32GB of GPU memory and NVLink.
Ascend A100
The NVIDIA A100 "Ampere" GPU, with a compute capability of 8.0, empowers advanced deep learning and scientific computing tasks. For instance, it accelerates and enhances the training of deep neural networks, enabling the training of intricate models like GPT-4 in significantly less time when compared to earlier GPU architectures.
The A100 comes equipped with 80GB of memory. here are 4 GPUs with NVLink, offering 320GB of usable GPU memory per node.
Examples
There are example jobs and code at GitHub