Samuel Shepard, Ph.D., a researcher in the University of Toledo bioinformatics lab of Associate Professor Alexei Fedorov, recently developed an algorithm for the prediction of certain genomic sequences, known as exons and introns, using midrange sequences of 20-50 nucleotides in length. These genomic patterns are said to display a non-random clustering of bases referred to as “mid-range inhomogeneity,” or MRI. Shepard hypothesized that the MRI patterns were different for exons and introns and would serve as a reliable predictor.
“We based our approach on Markov chain models, which are the basis for many gene prediction programs,” Shepard explained. “During the project, our algorithm read 12 million nucleotides of exons and introns each, and three million each were used to test the predictions.”
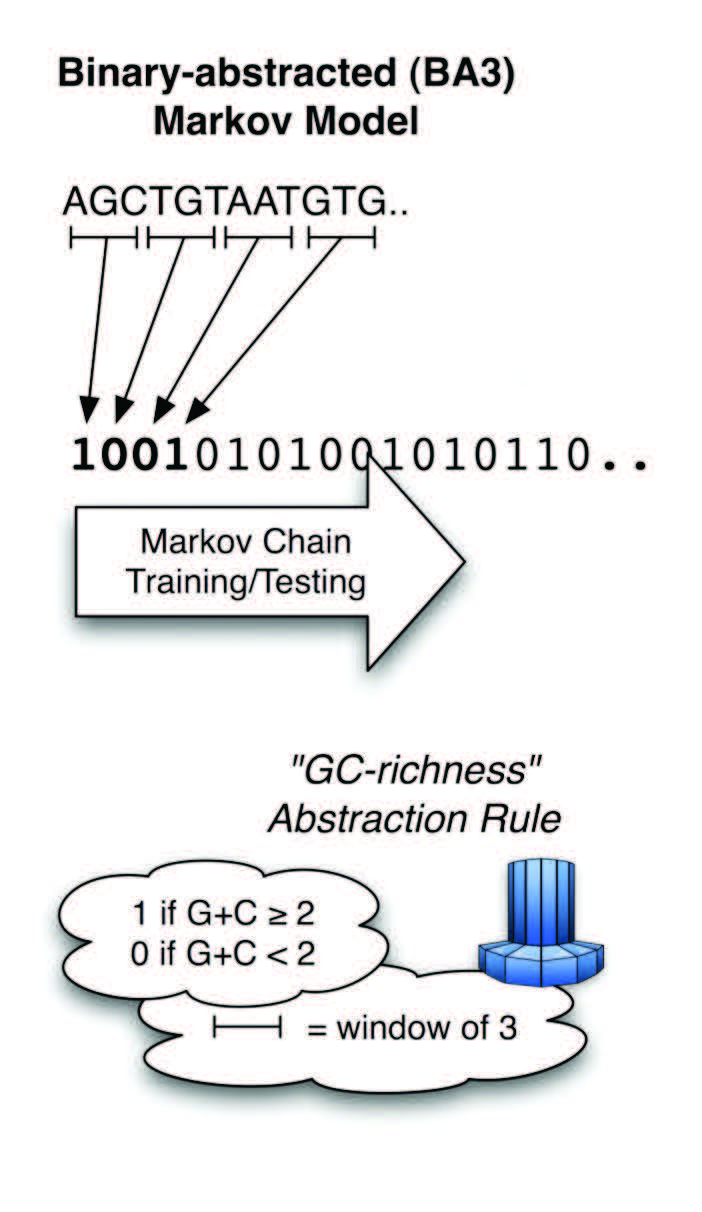
To circumvent the limitations of traditional Markov models, Shepard developed a technique known as binary-abstracted Markov modeling (BAMM). The procedure reduces mountains of nucleotide information into a much smaller binary code. Shepard tested abstraction rules for sequences of one or two nucleotides locally, but as larger sequences were studied, the possible abstraction outcomes increased exponentially.
Requiring far more computational horsepower, Shepard and his colleagues accessed the Ohio Supercomputer Center’s Glenn Cluster to optimize the abstraction process by using “hill-climbing” techniques that determine a single, maximal value for each abstraction space, rather than each of its possible values. Shepard and his team then combined different abstraction models to achieve an exon-intron prediction accuracy of greater than 95 percent.
--
Project lead: Samuel S. Shepard, University of Toledo
Research title: The characterization and utilization of middle-range sequence patterns
within the human genome
Funding source: National Science Foundation