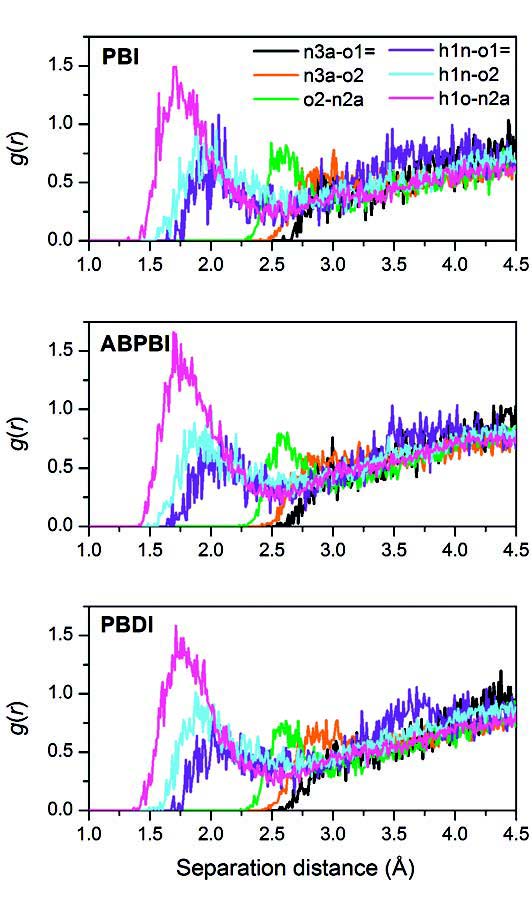
above: University of Cincinnati's Shuo Li used Ohio Supercomputer Center systems to create radial distribution function plots for various intermolecular atom pairs in three phosphoric acid-doped polybenzimidazoles: poly(2,2' -m-phenylene-5,5' -bibenzimidazole) (PBI), poly(2,5-benzimidazole) (ABPBI), and poly(p-phenylene benzobisimidazole) (PBDI).
The overarching driver for the development of fuel-cell power is its potential to provide clean, highly efficient power generation. A fuel cell produces electricity from fuel (on the anode side) and an oxidant (on the cathode side), which react in the presence of electrolytes, substances containing free ions that make the substance electrically conductive.
In one basic design, proton exchange membrane (PEM) fuel cells operate with a polymer electrolyte in the form of a thin, permeable sheet. Research led by Joel Fried, Ph.D., professor emeritus at the University of Cincinnati and Wright Brothers Institute endowed chair in nanomaterials at the University of Dayton, leveraged Ohio Supercomputer Center resources to investigate the potential of PEMs that use phosphoric acid (PA) to dope polybenzimidazole (PBI) for improved performance. PBI is a polymer with an extremely high melting point used to fabricate protective apparel, such as firefighter coats, astronaut space suits and high-temperature protective gloves. Fuel cells using PA-doped PBI can operate in the range of 150-200 degrees Celsius.
“Compared with conventional PEM membranes (below 100 degrees Celsius), the high-temperature operation of a PA-doped PBI membrane allows increased catalyst activity at the electrodes and better carbon-monoxide tolerance in hydrogen fuel,” said Shuo Li, a former Ph.D. candidate in chemical engineering advised by Fried at the University of Cincinnati. “Ultimate success in the continuing development of these PEMs requires a fundamental understanding of the molecular structure of the membrane and the diffusional process of protons and small molecules.”
Proton transfer in fuel-cell membranes consists of proton-hopping along the hydrogen bond network (measured in picoseconds) and the diffusion of proton carriers between hops (measured in nanoseconds). Due to different time scales and difficult experimental techniques, Li explained, multi-scale simulation techniques are usually required to study these properties. Multi-scale modeling using quantum mechanics, molecular dynamics, and Atom-centered Density Matrix Propagation ab initio molecular dynamics were employed to study proton transfer and the hydrogen bond network structure in PA-doped PBI membranes.
Quantum mechanics calculations of gas-phase proton affinity, interaction energy and energy barriers for different proton transfer pathways indicated that proton transfer is prone to occur between the same molecules or between a molecule and its corresponding ion. The influences of different PBI structures, PA-doping levels, temperatures and water contents were evaluated through molecular dynamics simulations of hydrogen bond network structures and vehicular diffusion properties in neat, hydrated and PA-doped PBIs. Calculations were performed on the PA-doped PBI model to study the initial steps of proton transfer and the interfacial properties between PBI and PA.
--
Project lead: Joel Fried, University of Dayton
Research title: Computational chemistry and molecular simulations of phosphoric acid-doped polybenzimidazole
Funding source: American Chemical Society
Web site: www2.udayton.edu/engineering/profiles/fried_joel.php