
By combining state-of-the-art computational methods with theoretical considerations, a research group led by Maryam Ghazisaeidi at The Ohio State University is studying the connection between microscopic physical phenomena and macroscopic mechanical behavior of engineering materials.
“Our research is at the intersection of materials science, physics and mechanics,” said Ghazisaeidi, Ph.D., an associate professor in the materials science and engineering department. “We use atomic-scale computations—electronic structures and classical potentials—coupled to larger length-scale continuum and statistical mechanics to improve and predict the properties of existing and new materials.”
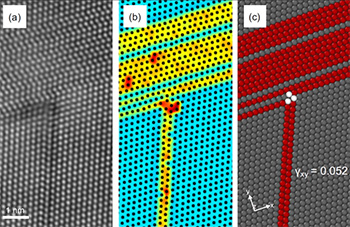
Recently, Ghazisaeidi’s group undertook a project to better understand the remarkable mechanical properties of high entropy alloys (HEAs) by investigating their fundamental deformation mechanisms—micro-scale processes, such as dislocation and stacking fault structures and energies, that allow it to change its internal structure, shape or volume. HEAs are formed at the atomic level of equal or near-equal concentrations of multiple metallic elements, crystalize as solid solutions and don’t take a secondary form, or phase, such as a gas or liquid.
The Ghazisaeidi group is leveraging Ohio Supercomputer Center (OSC) systems to model these attributes in concentrated multielement alloys (CMAs), of which the singlephase HEAs are an interesting subset. CMAs are fundamentally different from those in dilute alloys—those with a much more varied concentration of constituent elements. The group also looks to develop new computational tools that enable dislocation modeling in CMAs with the quantum mechanical modelling method known as density functional theory (DFT). The group employs the DFT code VASP—currently the most widely used plane-wave, first-principles code, to perform the required calculations.
“Electronic structure calculations of dislocations typically require larger simulation sizes and accurate treatments of boundary conditions,” Ghazisaeidi noted. “In this project, we take a first-principles computational approach using recently-developed capabilities to address two questions concerning these multicomponent alloys: What is the correct way of thinking about structural defects? What is the solid-solution strengthening mechanism?”
To answer these questions, Ghazisaeidi and her team are logging substantial computation time on OSC’s Ruby and Owens Clusters, as well as leveraging the Center’s high performance storage resources.
“Existing approaches to modeling material behavior generally rely on phenomenological constitutive relations that must be calibrated experimentally, and therefore lack predictive capability,” Ghazisaeidi said. “Connecting the accurate atomic-scale study of deformation to the overall mechanical properties will potentially transform alloy design by replacing trial-and-error approaches with quantitative predictive models.”
_________
PROJECT LEAD // Maryam Ghazisaeidi, Ph.D., The Ohio State University
RESEARCH TITLE // Understanding novel characteristics of defects in concentrated solid solutions from first principles calculations
FUNDING SOURCE // National Science Foundation
WEBSITE // ghazisaeidigroup.engineering.osu.edu